Enfermedad de Huntington
Perspectiva general
La enfermedad de Huntington es una enfermedad hereditaria poco frecuente que provoca la degradación progresiva (degeneración) de las células nerviosas del cerebro. La enfermedad de Huntington tiene una amplia repercusión en las capacidades funcionales de una persona y, generalmente, ocasiona trastornos del movimiento, el pensamiento (cognitivos) y psiquiátricos.
Los síntomas de la enfermedad de Huntington pueden presentarse en cualquier momento, pero a menudo aparecen por primera vez cuando las personas tienen entre 30 y 50 años. Si la afección se presenta antes de los 20 años, se llama enfermedad de Huntington juvenil. Cuando la enfermedad de Huntington se presenta de manera temprana, los síntomas son algo diferentes y es posible que la enfermedad puede progrese más rápido.
Hay medicamentos disponibles para ayudar a controlar los síntomas de la enfermedad de Huntington. Sin embargo, los tratamientos no pueden prevenir el deterioro físico, mental y de comportamiento asociado a la afección.
Síntomas
La enfermedad de Huntington a menudo ocasiona trastornos motrices, cognitivos y psiquiátricos con una amplia gama de signos y síntomas. Los síntomas que se presentan al principio varían mucho de una persona a otra. Algunos síntomas parecen ser más dominantes o tienen un mayor efecto sobre la capacidad funcional, pero eso puede cambiar durante el curso de la enfermedad.
Trastornos del movimiento
Los trastornos del movimiento asociados a la enfermedad de Huntington pueden incluir problemas de movimiento involuntario y deterioro de los movimientos voluntarios, por ejemplo:
- Movimientos espasmódicos o de contorsión involuntarios (corea)
- Problemas musculares, como rigidez o contracturas musculares (distonía)
- Movimientos oculares lentos o inusuales
- Alteración en la marcha, la postura y el equilibrio
- Dificultad para hablar o tragar
El deterioro de los movimientos voluntarios, a diferencia de los movimientos involuntarios, puede tener una mayor repercusión en la capacidad de la persona para trabajar, hacer actividades cotidianas, comunicarse y ser independiente.
Trastornos cognitivos
Algunos de los signos del deterioro cognitivo generalmente relacionados a la enfermedad de Huntington son los siguientes:
- Dificultad para organizarse, establecer prioridades o enfocarse en tareas
- Falta de flexibilidad o tendencia a quedarse atascado en un pensamiento, conducta o acción (perseveración)
- Falta de control de los impulsos, que puede tener como consecuencia arrebatos, actuar sin pensar y promiscuidad sexual
- Falta de conciencia sobre las conductas y aptitudes propias
- Lentitud para procesar pensamientos o encontrar las palabras indicadas
- Dificultad para aprender información nueva
Trastornos psiquiátricos
El trastorno psiquiátrico más frecuente asociado a la enfermedad de Huntington es la depresión. Y no se trata solamente de una reacción al recibir el diagnóstico de enfermedad de Huntington. Por el contrario, la depresión parece ocurrir debido a lesiones en el cerebro y posteriores cambios en el funcionamiento cerebral. Estos son algunos de los signos y síntomas:
- Sensación de irritabilidad, tristeza o apatía
- Aislamiento social
- Insomnio
- Fatiga y pérdida de energía
- Ideas frecuentes sobre la muerte, morir o el suicidio
Otros trastornos psiquiátricos frecuentes son:
- Trastorno obsesivo compulsivo, una afección caracterizada por pensamientos recurrentes e invasivos, y conductas repetitivas
- Manía, que puede ocasionar un estado de ánimo elevado, hiperactividad, conductas impulsivas y autoestima excesiva
- Trastorno bipolar, una afección con episodios alternados de depresión y manía
Además de los trastornos mencionados, es común que las personas que padecen la enfermedad de Huntington sufran pérdida de peso, especialmente a medida que la enfermedad avanza.
Síntomas de la enfermedad de Huntington juvenil
El inicio y el avance de la enfermedad de Huntington en personas jóvenes pueden ser ligeramente diferente a los de los adultos. Entre los problemas que suelen presentarse al principio del curso de la enfermedad se incluyen los siguientes:
Cambios en la conducta
- Dificultad para prestar atención
- Disminución rápida y significativa del desempeño escolar general
- Problemas de conducta
Cambios físicos
- Músculos contraídos y rígidos que afectan la marcha (especialmente en los niños pequeños)
- Temblores o movimientos involuntarios leves
- Caídas frecuentes o torpeza
- Convulsiones
Cuándo consultar al médico
Consulta con el proveedor de atención médica si observas cambios en tus movimientos, estado emocional o capacidad mental. Los signos y síntomas de la enfermedad de Huntington pueden deberse a varias afecciones diferentes. Por lo tanto, es importante obtener un diagnóstico completo y rápido.
Causas
La enfermedad de Huntington se produce a causa de una diferencia heredada en un solo gen. La enfermedad de Huntington es un trastorno autosómico dominante, lo que significa que una persona necesita solo una copia del gen atípico para desarrollar el trastorno.
A excepción de los cromosomas sexuales, una persona hereda dos copias de cada gen, una copia de cada padre. Un padre con un gen atípico podría trasmitir la copia atípica del gen o la copia sana. Por lo tanto, cada hijo tiene un 50 % de probabilidades de heredar el gen que causa el trastorno genético.
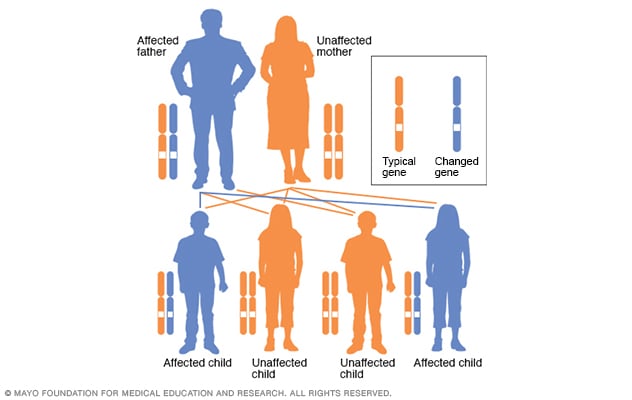
En un trastorno autosómico dominante, el gen alterado es un gen dominante. Está ubicado en uno de los autosomas, que son los cromosomas no sexuales. Solo se necesita un gen alterado para que una persona se vea afectada por este tipo de afección. Una persona con una afección autosómica dominante, en este caso, el padre, tiene un 50 % de probabilidades de tener un hijo con un gen alterado. Del mismo modo, tiene un 50 % de probabilidades de tener un hijo que no se vea afectado.
Complicaciones
Luego de la aparición de la enfermedad de Huntington, las capacidades funcionales de una persona empeoran progresivamente con el tiempo. La tasa de avance y duración de la enfermedad varía. El tiempo desde la aparición de los primeros síntomas hasta la muerte por lo general es de 10 a 30 años. La enfermedad de Huntington juvenil generalmente provoca la muerte en un plazo de 10 años después de la aparición de los síntomas.
La depresión clínica relacionada con la enfermedad de Huntington puede aumentar el riesgo de suicidio. Algunas investigaciones sugieren que el mayor riesgo de suicidio se da antes de que se realice el diagnóstico y en las etapas intermedias de la enfermedad, cuando una persona comienza a perder independencia.
Eventualmente, una persona que tiene la enfermedad de Huntington requerirá ayuda con todas las actividades cotidianas y los cuidados médicos. En las etapas finales de la enfermedad, la persona probablemente quede postrada en una cama y sin poder hablar. Por lo general, una persona con la enfermedad de Huntington puede comprender lo que se dice y reconoce a sus amigos y familiares; sin embargo, otras personas no reconocerán a sus familiares.
Las causas frecuentes de muerte comprenden las siguientes:
- Neumonía u otras infecciones
- Lesiones relacionadas con caídas
- Complicaciones relacionadas con la imposibilidad de tragar
Prevención
A las personas que tienen antecedentes familiares conocidos de enfermedad de Huntington les preocupa saber si les transmitirán el gen de Huntington a sus hijos. Estas personas pueden considerar pruebas genéticas y opciones de planificación familiar.
Si un progenitor que está en riesgo considera realizarse pruebas genéticas, puede ser útil programar una cita con un consejero genético. El consejero genético conversará contigo sobre los posibles riesgos de un resultado positivo de la prueba, que indicaría que el progenitor desarrollará la enfermedad. Además, las parejas deberán tomar decisiones adicionales con respecto a si tendrán hijos o si tendrán en cuenta otras alternativas, como análisis prenatales para detectar el gen o fertilización in vitro con esperma u óvulos de donantes.
Otra opción para las parejas es la fertilización in vitro y el diagnóstico genético preimplantacional. En este proceso, se extraen los óvulos de los ovarios y se fecundan con el esperma del padre en un laboratorio. Se analizan los embriones para detectar la existencia del gen de Huntington, y se implantan en el útero de la madre solo los que hayan dado negativo en el análisis del gen de Huntington.
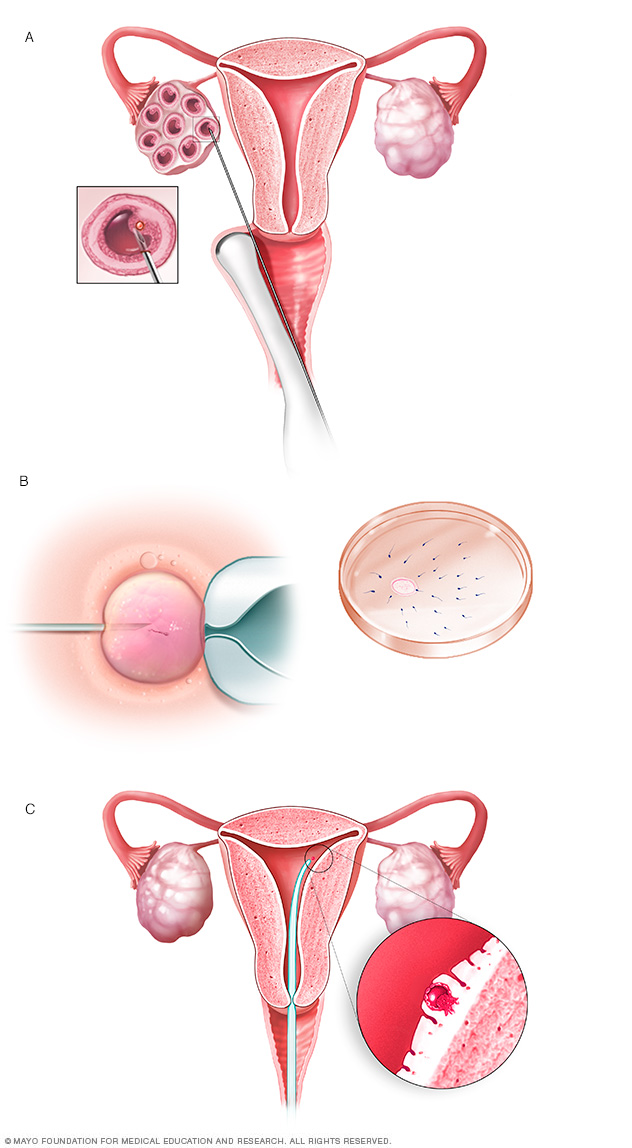
Durante la fertilización in vitro, se extraen los óvulos de unos sacos, llamados folículos, dentro del ovario (A). El óvulo se fertiliza al inyectar un solo espermatozoide dentro del óvulo o al mezclar el óvulo con el esperma en una placa de Petri (B). El óvulo fecundado, que también se conoce como embrión, se transfiere al interior del útero (C).
Diagnóstico
El diagnóstico preliminar de la enfermedad de Huntington se basa principalmente en tus respuestas a preguntas, en una exploración física general, una revisión de la historia clínica familiar y exámenes neurológicos y psiquiátricos.
Examen neurológico
El neurólogo te hará preguntas y te hará pruebas relativamente simples:
- Síntomas motores, como los reflejos, la fuerza muscular y el equilibrio
- Síntomas sensoriales, incluido el sentido del tacto, la visión y la audición
- Síntomas psiquiátricos, como el estado de ánimo y mental
Análisis neuropsicológico
El neurólogo también puede realizar pruebas estandarizadas para evaluar lo siguiente:
- Memoria
- Razonamiento
- Agilidad mental
- Conocimientos de idiomas
- Razonamiento espacial
Evaluación psiquiátrica
Es probable que se te remita a un psiquiatra para que te examine e intente detectar una serie de factores que podrían contribuir a tu diagnóstico, entre ellos:
- Estado emocional
- Patrones de comportamiento
- Calidad de juicio
- Capacidad de afrontar una situación difícil
- Signos de trastornos de pensamiento
- Evidencia de toxicomanía
Pruebas por imágenes del cerebro y de su actividad
El proveedor de atención médica puede pedir pruebas por imágenes del cerebro para evaluar su estructura o su actividad. Las tecnologías de obtención de imágenes pueden incluir resonancias magnéticas o tomografías computarizadas que muestran imágenes detalladas del cerebro.
Estas imágenes pueden revelar cambios en el cerebro en las áreas afectadas por la enfermedad de Huntington. Es posible que estos cambios no aparezcan al principio del curso de la enfermedad. Estas pruebas también se pueden usar para descartar otras afecciones que podrían estar provocando los síntomas.
Asesoramiento en genética y análisis
Si los síntomas indican claramente que se trata de la enfermedad de Huntington, el proveedor de atención médica te recomendará hacerte una prueba genética para identificar el gen atípico.
Esta prueba puede confirmar el diagnóstico. También puede resultar muy útil si no tienes antecedentes familiares conocidos de la enfermedad de Huntington o si no se confirmó el diagnóstico de otro familiar con una prueba genética. Sin embargo, la prueba no proporcionará información para determinar un plan de tratamiento.
Antes de hacerte esta prueba, el consejero genético te explicará las ventajas y las desventajas de conocer los resultados. El consejero genético también puede responder preguntas sobre el tipo de trasmisión hereditaria de la enfermedad de Huntington.
Análisis genético predictivo
Puedes someterte a una prueba genética si tienes antecedentes familiares de la enfermedad, pero no tienes síntomas. A esto se lo conoce como análisis predictivo. La prueba no puede determinar cuándo comenzará la enfermedad o qué síntomas aparecerán primero.
Algunas personas pueden decidir hacerse la prueba porque les resulta más estresante no tener conocimiento. Otras pueden preferir hacerse el análisis antes de tener hijos.
Los riesgos pueden comprender problemas con el seguro o con el empleo futuro y el estrés de enfrentar una enfermedad mortal. En principio, existen leyes federales que prohíben el uso de la información de las pruebas genéticas para discriminar a las personas con enfermedades genéticas.
Estas pruebas solo se hacen luego de una consulta con un consejero genético.
Tratamiento
Ningún tratamiento puede alterar el curso de la enfermedad de Huntington, pero los medicamentos pueden aliviar algunos síntomas de los trastornos psiquiátricos y del movimiento. Además, múltiples intervenciones pueden ayudar a una persona a adaptarse a los cambios en sus capacidades durante un cierto tiempo.
Es probable que los medicamentos evolucionen en el curso de la enfermedad, dependiendo de los objetivos generales del tratamiento. Además, los medicamentos que tratan algunos síntomas pueden producir efectos secundarios que empeoran otros síntomas. Los objetivos del tratamiento se revisarán y actualizarán periódicamente.
Medicamentos para los trastornos del movimiento
Los medicamentos para tratar los trastornos del movimiento incluyen:
- Los medicamentos para controlar el movimiento incluyen la tetrabenazina (Xenazine) y la deutetrabenazina (Austedo), que están aprobadas específicamente por la Administración de Alimentos y Medicamentos de Estados Unidos para reducir los movimientos espasmódicos y de contorsión involuntarios (corea) asociados con la enfermedad de Huntington. Sin embargo, estos medicamentos no tienen ningún efecto en la progresión de la enfermedad. Los posibles efectos secundarios incluyen somnolencia, agitación y el riesgo de empeorar o desencadenar la depresión u otras afecciones psiquiátricas.
-
Los medicamentos antipsicóticos, como el haloperidol y la flufenacina, tienen el efecto secundario de suprimir los movimientos. Por lo tanto, pueden ser beneficiosos para tratar la corea. Sin embargo, estos medicamentos pueden empeorar las contracciones involuntarias (distonía), la agitación y la somnolencia.
Si bien otros medicamentos, como la olanzapina (Zyprexa) y el aripiprazol (Abilify), pueden tener menos efectos secundarios, deben usarse con cuidado, ya que también pueden empeorar los síntomas.
- Otros medicamentos que pueden ayudar a reducir la corea incluyen la amantadina (Gocovri, Osmolex ER), el levetiracetam (Keppra, Elepsia XR, Spritam) y el clonazepam (Klonopin). Sin embargo, los efectos secundarios pueden limitar su uso.
Medicamentos para trastornos psiquiátricos
Los medicamentos para tratar los trastornos psiquiátricos variarán según los trastornos y los síntomas. Los posibles tratamientos incluyen lo siguiente:
- Los antidepresivos incluyen medicamentos como citalopram (Celexa), escitalopram (Lexapro), fluoxetina (Prozac) y sertralina (Zoloft). Estos medicamentos también pueden tener efectos en el tratamiento del trastorno obsesivo compulsivo. Los efectos secundarios pueden incluir náuseas, diarrea, somnolencia y presión arterial baja.
- Los medicamentos antipsicóticos, como la quetiapina (Seroquel) y olanzapina (Zyprexa), pueden inhibir los ataques de violencia, la agitación y otros síntomas de los trastornos del estado de ánimo o la psicosis. Sin embargo, estos mismos medicamentos pueden provocar diferentes trastornos del movimiento.
- Los medicamentos estabilizadores del estado de ánimo, que pueden ayudar a evitar los altibajos emocionales asociados con el trastorno bipolar, incluyen anticonvulsivos, como el divalproex (Depakote), la carbamazepina (Tegretol, Carbatrol, Epitol, otros) y la lamotrigina (Lamictal).
Psicoterapia
Un psicoterapeuta, un psiquiatra, psicólogo o trabajador social clínico, puede proporcionar terapia de conversación para ayudar con los problemas de comportamiento, desarrollar estrategias de afrontamiento, manejar las expectativas durante la progresión de la enfermedad y ayudar a los miembros de la familia a comunicarse.
Terapia del habla
La enfermedad de Huntington puede afectar significativamente el control de los músculos de la boca y la garganta que son esenciales para hablar, comer y tragar. Un logopeda puede ayudar a mejorar tu capacidad de hablar claramente o te puede enseñar a utilizar los dispositivos de comunicación, como por ejemplo una pizarra que exponga imágenes de artículos y actividades diarias. Los logopedas también pueden tratar las dificultades con los músculos que se utilizan para comer y tragar.
Fisioterapia
Un fisioterapeuta puede enseñarte ejercicios adecuados y seguros para aumentar tu fuerza, flexibilidad, equilibrio y coordinación. Estos ejercicios pueden ayudarte a conservar el rango de movimiento el mayor tiempo posible y pueden reducir el riesgo de caídas.
Las instrucciones sobre una postura adecuada y el uso de apoyos para mejorarla pueden ayudar a reducir la gravedad de algunos problemas de movimiento.
Si necesitas usar un andador o una silla de ruedas, el fisioterapeuta puede brindarte instrucciones sobre su uso adecuado y la postura correcta al usarlos. Además, las rutinas de ejercicios se pueden adaptar al nuevo nivel de movimiento.
Terapia ocupacional
Un terapeuta ocupacional puede ayudar a la persona con enfermedad de Huntington, a sus familiares y a los cuidadores con los dispositivos de asistencia que mejoran las capacidades funcionales. Estas estrategias pueden ser las siguientes:
- Pasamanos en el hogar
- Dispositivos de asistencia para realizar ciertas actividades, como bañarse y vestirse
- Utensilios para comer y beber adaptados a las personas con motricidad fina limitada
Estilo de vida y remedios caseros
Controlar la enfermedad de Huntington afecta a la persona con el trastorno, a los miembros de su familia y a otras personas encargadas del cuidado en casa. A medida que la enfermedad avanza, la persona dependerá cada vez más de las personas encargadas del cuidado. Se deberán abordar diversos asuntos, y las estrategias para afrontarlos irán cambiando a lo largo del tiempo.
Alimentación y nutrición
Los factores relacionados con la alimentación y nutrición comprenden los siguientes:
- Dificultad para mantener un peso corporal saludable. La dificultad para alimentarse, las necesidades calóricas mayores debidas al esfuerzo físico o los problemas metabólicos desconocidos pueden ser la causa. Para obtener una nutrición apropiada, es posible que debas comer más de tres comidas al día o usar suplementos alimentarios.
- Dificultad para masticar, tragar y con la motricidad fina. Estos problemas pueden limitar la cantidad de comida que se consume y aumentar el riesgo de atragantamiento. Puede ser útil eliminar las distracciones durante una comida y elegir alimentos que sean más fáciles de comer. Los utensilios diseñados para personas con habilidades de motricidad fina limitadas y los vasos con tapa y popote (sorbete), o los vasos con pico, también pueden ser útiles.
Con el tiempo, una persona que tiene la enfermedad de Huntington necesitará ayuda para comer y beber.
Cómo controlar los trastornos psiquiátricos y cognitivos
Los familiares y las personas encargadas del cuidado pueden crear un ambiente que ayude a una persona que tiene la enfermedad de Huntington a evitar el estrés y a controlar los trastornos cognitivos y de conducta. Estas estrategias comprenden las siguientes:
- Usar calendarios y horarios para mantener una rutina habitual
- Iniciar las tareas con recordatorios o asistencia
- Priorizar u organizar el trabajo o las actividades
- Dividir las tareas en pasos razonables
- Crear un ambiente lo más calmo, simple y estructurado posible
- Identificar y evitar factores de estrés que puedan provocar arrebatos, irritabilidad, depresión u otros problemas
- En el caso de niños en edad escolar o adolescentes, hablar con el personal de la escuela para elaborar un plan educativo individualizado adecuado
- Darle a la persona oportunidades para que mantenga interacciones sociales y amistades en la mayor medida posible
Estrategias de afrontamiento, y apoyo
A number of strategies may help people with Huntington's disease and their families cope with the challenges of the disease.
Servicios de apoyo
Los servicios de apoyo para las personas con la enfermedad de Huntington y sus familias comprenden lo siguiente:
- Organismos sin fines de lucro, como la Huntington's Disease Society of America (Sociedad Estadounidense de la Enfermedad de Huntington), que brindan educación a las personas encargadas del cuidado, derivaciones a servicios externos y grupos de apoyo para las personas que viven con la enfermedad y las personas encargadas de su cuidado.
- Oficinas de servicios sociales o de salud locales y estatales, que pueden ofrecer atención médica durante el día a las personas que viven con la enfermedad, programas de asistencia de comidas o servicios de relevo para las personas encargadas del cuidado.
Planificación para la atención residencial y terminal
Como la enfermedad de Huntington provoca la pérdida progresiva de la capacidad funcional y la muerte, es importante prever los aspectos relacionados con la atención médica que necesitará el paciente en las etapas avanzadas de la enfermedad y cerca del final de su vida. Hablar a tiempo sobre este tipo de atención médica le permite a la persona con la enfermedad de Huntington participar en la toma de decisiones y expresar sus deseos relacionados con la atención médica.
Crear los documentos legales que definen las instrucciones relacionadas con la atención a pacientes terminales puede ser útil para todos. Con estos documentos, se empodera a la persona que tiene la enfermedad y se evitan conflictos familiares cuando la enfermedad ya está muy avanzada. El proveedor de atención médica puede explicarte las ventajas y las desventajas de las opciones de atención médica en el momento en el que se pueden analizar minuciosamente todas las opciones.
Algunos de los asuntos que se deben tener en cuenta son los siguientes:
- Centros de atención médica. Es probable que, en las etapas avanzadas de la enfermedad, el paciente necesite cuidados de enfermería en el hogar o atención en un centro de vivienda tutelada o en un asilo de ancianos y convalecientes.
- Cuidado para pacientes terminales. Los servicios de atención paliativa brindan atención con el menor malestar posible durante la etapa final de la vida de una persona que se acerca al momento de la muerte. Este tipo de atención también brinda apoyo y educación a los familiares para ayudarles a comprender el proceso de la muerte.
- Testamentos en vida. Los testamentos en vida son documentos legales que le permiten a una persona explicar en detalle sus preferencias de atención médica para cuando ya no pueda tomar decisiones. Por ejemplo, en estas instrucciones, la persona puede indicar si quiere o no que le hagan intervenciones para mantenerla con vida o recibir un tratamiento agresivo para combatir una infección.
- Directrices médicas anticipadas. Con estos documentos legales, el paciente puede nombrar a una o más personas para que tomen decisiones en su nombre. Se pueden crear directrices médicas anticipadas para las cuestiones relacionadas con las decisiones médicas o con los asuntos financieros.
Preparación antes de la cita
Si presentas algún signo o síntoma relacionado con la enfermedad de Huntington es probable que, luego de una consulta inicial, tu proveedor de atención médica te remita al neurólogo.
Para la evaluación clínica de un posible trastorno neurológico, puede ser importante revisar tus síntomas, estado mental, y antecedentes médicos personales y familiares.
Qué puedes hacer
Antes de la cita, prepara una lista que incluya lo siguiente:
- Signos o síntomas, o cualquier cambio que te resulte fuera de lo normal, que puedan causarte inquietudes
- Cambios recientes o situaciones de estrés en tu vida
- Todos los medicamentos, incluidos los medicamentos de venta sin receta y los suplementos alimentarios, y sus dosis
- Antecedentes familiares de enfermedad de Huntington u otros trastornos que puedan ocasionar problemas motrices o afecciones psiquiátricas
Puedes pedirle algún amigo o familiar que te acompañe a la cita. Esta persona puede brindarte apoyo y ofrecer una perspectiva diferente sobre el efecto de los síntomas en tus capacidades funcionales.
Qué esperar del médico
Es probable que el proveedor de atención médica te haga algunas preguntas, incluidas las siguientes:
- ¿Cuándo comenzaste a tener síntomas?
- ¿Los síntomas han sido continuos o intermitentes?
- ¿A alguien de tu familia le diagnosticaron la enfermedad de Huntington alguna vez?
- ¿A ti o a alguien de tu familia se les diagnosticó otro trastorno motriz o psiquiátrico?
- ¿Tienes problemas para realizar tu trabajo, tus tareas escolares o tus actividades diarias?
- ¿Alguna persona de tu familia murió joven?
- ¿Alguna persona de tu familia se encuentra en un asilo de ancianos y convalecientes?
- ¿Alguna persona de tu familia es inquieta o se mueve todo el tiempo?
- ¿Has notado algún cambio en tu estado de ánimo general?
- ¿Te sientes triste todo el tiempo?
- ¿Has pensado alguna vez en el suicidio?
Last Updated Jul 19, 2022
© 2024 Mayo Foundation for Medical Education and Research (MFMER). All rights reserved. Terms of Use