Enfermedad de Wilson
Perspectiva general
La enfermedad de Wilson es una afección hereditaria poco frecuente que causa la acumulación de cobre en varios órganos, especialmente el hígado, el cerebro y los ojos. A la mayoría de las personas con la enfermedad de Wilson, esta se les diagnostica entre los 5 y los 35 años. Pero también pueden verse afectadas personas de menor y mayor edad.
El cobre desempeña una función importante en el desarrollo de nervios saludables, huesos, colágeno y melanina, la pigmentación de la piel. Por lo general, consumes cobre en los alimentos que comes. El hígado produce una sustancia llamada bilis que elimina todo el cobre sobrante.
Sin embargo, las personas con la enfermedad de Wilson no pueden eliminar el cobre de manera adecuada, por lo que se acumula. Muchas veces, esta afección puede ser mortal si no se trata. Cuando se diagnostica pronto, la enfermedad de Wilson es tratable, y muchas personas que la padecen llevan una vida normal.
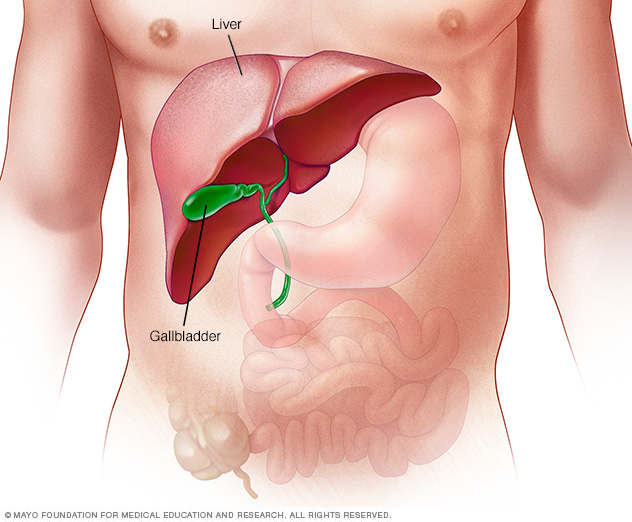
El hígado es el órgano interno más grande del cuerpo. Es casi del tamaño de una pelota de fútbol. Se ubica principalmente en la parte superior derecha de la zona del estómago, por encima de este.
Síntomas
La enfermedad de Wilson se presenta al nacer, pero los síntomas no aparecen hasta que los niveles de cobre se acumulan en el cerebro, el hígado, los ojos u otro órgano. Los síntomas varían según las partes del cuerpo afectadas por la enfermedad.
Estos síntomas son los siguientes:
- Cansancio y pérdida del apetito
- Color amarillento de la piel y la parte blanca de los ojos, que se denomina ictericia
- Anillos de color marrón dorado o cobrizo alrededor del iris de los ojos, conocidos como anillos de Kayser-Fleischer
- Acumulación de líquido en las piernas o el abdomen
- Problemas para hablar, para tragar o con la coordinación física
- Depresión, cambios en el estado de ánimo y en la personalidad
- Dificultad para conciliar el sueño y permanecer dormido
- Movimientos descontrolados o rigidez muscular
Cuándo consultar con un médico
Programa una cita con el médico o con otro proveedor principal de atención médica si tienes síntomas que te preocupen, en especial, si un miembro de tu familia tiene la enfermedad de Wilson.
Causas
La causa de la enfermedad de Wilson es un gen alterado que se transmite de padres a hijos. Si solo tienes un gen afectado, no contraerás la enfermedad, pero serás portador. Esto significa que podrías pasar el gen afectado a tus hijos.
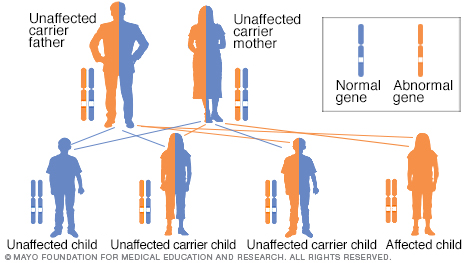
La causa de la enfermedad de Wilson es un gen alterado que se hereda del padre y la madre. Heredar un gen alterado de un solo progenitor rara vez afecta la salud de una persona. Sin embargo, esa persona tiene un gen alterado y un gen normal. Dos personas portadoras de genes alterados tienen un 25 % de probabilidades de tener un hijo con dos genes normales, un 50 % de probabilidades de tener un hijo portador y un 25 % de probabilidades de tener un hijo afectado por la enfermedad de Wilson.
Factores de riesgo
Si tus padres o hermanos tienen la enfermedad de Wilson, es posible que afrontes un riesgo mayor de padecer esta enfermedad. Pregúntale al médico si deben realizarte un análisis genético para determinar si tienes la enfermedad de Wilson. Diagnosticar la enfermedad con la mayor antelación posible aumenta drásticamente las posibilidades de que el tratamiento sea exitoso.
Complicaciones
Si la enfermedad de Wilson no se trata, puede causar la muerte en algunas ocasiones. Las complicaciones graves incluyen las siguientes:
- Cirrosis, que es la formación de cicatrices en el hígado. Cuando las células hepáticas intentan reparar los daños causados por los niveles altos de cobre, se forma tejido cicatricial en el hígado. Esto dificulta su funcionamiento.
- Insuficiencia hepática. Esta afección puede ocurrir repentinamente, lo que se conoce como insuficiencia hepática aguda o enfermedad de Wilson descompensada. También puede desarrollarse lentamente a lo largo de los años. El trasplante de hígado puede ser una opción de tratamiento.
- Problemas duraderos en el sistema nervioso. Los temblores, los movimientos musculares involuntarios, la torpeza al andar y la dificultad para hablar suelen mejorar con el tratamiento de la enfermedad de Wilson. Sin embargo, algunas personas tienen problemas duraderos en el sistema nervioso, incluso con el tratamiento.
- Problemas renales. La enfermedad de Wilson puede dañar los riñones, lo que ocasiona problemas como cálculos renales y una cantidad anormal de aminoácidos eliminados en la orina.
- Problemas de salud mental. Pueden ser cambios en la personalidad, depresión, irritabilidad, trastorno bipolar o psicosis.
- Problemas sanguíneos. Pueden incluir la destrucción de glóbulos rojos, lo que se conoce como hemólisis, que deriva en anemia e ictericia.
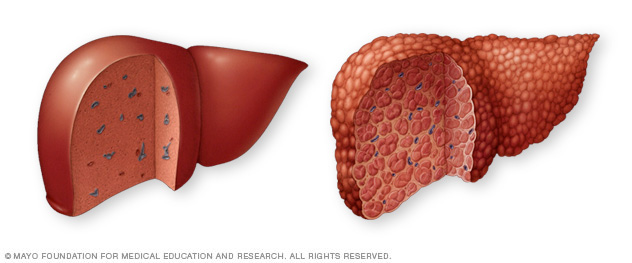
Un hígado sano, a la izquierda, no muestra signos de tejido fibroso. En un hígado con cirrosis, a la derecha, el tejido fibroso reemplaza el tejido hepático sano.
Diagnóstico
El diagnóstico de la enfermedad de Wilson puede ser difícil porque sus síntomas suelen parecerse a los de otras enfermedades hepáticas, como la hepatitis. Además, pueden aparecer con el tiempo. Los cambios en el comportamiento que aparecen progresivamente pueden ser especialmente difíciles de vincular con la enfermedad de Wilson.
Para hacer el diagnóstico, los médicos se basan en los síntomas y resultados de pruebas. Las pruebas y los procedimientos que se usan para diagnosticar la enfermedad de Wilson incluyen lo siguiente:
- Análisis de sangre y de orina. Los análisis de sangre pueden vigilar la función hepática y verificar el nivel de una proteína denominada ceruloplasmina que fija el cobre en la sangre. También pueden comprobar el nivel de cobre en sangre. También es posible que el médico quiera medir la cantidad de cobre eliminado en la orina durante un período de 24 horas.
- Examen ocular. Mediante un microscopio con luz de alta intensidad, un oftalmólogo comprueba si hay anillos de Kayser-Fleischer en los ojos. Esto se llama examen con lámpara de hendidura. Estos anillos se forman debido al exceso de cobre que hay en los ojos. La enfermedad de Wilson también está relacionada con un tipo de catarata, denominada catarata en girasol. Estas cataratas se pueden ver durante un examen ocular.
- Extracción de una muestra de tejido hepático para su análisis, lo que se conoce también como biopsia. En una biopsia, el médico inserta una aguja fina a través de la piel dentro del hígado. Luego, extrae una pequeña muestra de tejido. En el laboratorio, se analiza el tejido para detectar un exceso de cobre.
- Pruebas genéticas. Un análisis de sangre puede precisar los cambios genéticos que causan la enfermedad de Wilson. Si tienes el gen alterado que causa esta enfermedad, los médicos también pueden examinar a tus hermanos. Si alguno tiene dicho gen, ese hermano puede iniciar el tratamiento antes de que se presenten los síntomas.

En una biopsia de hígado, se extirpa una muestra pequeña de tejido hepático que se analiza en el laboratorio. Normalmente, la biopsia de hígado se hace al insertar una aguja delgada a través de la piel hasta llegar al hígado.
Tratamiento
Tu médico puede recomendarte unos medicamentos denominados quelantes del cobre. Estos medicamentos se adhieren al cobre y hacen que los órganos lo liberen en el torrente sanguíneo. Los riñones luego filtran el cobre y lo eliminan a través de la orina.
El tratamiento posterior consiste en evitar que el cobre vuelva a acumularse. En el caso de un daño hepático grave, puede ser necesario realizar un trasplante de hígado.
Medicamentos
Si tomas medicamentos para la enfermedad de Wilson, el tratamiento es de por vida. Estos medicamentos incluyen los siguientes:
- Penicilamina (Cuprimine, Depen). La penicilamina es un quelante del cobre. Puede causar efectos secundarios graves, como problemas en la piel y los riñones, y empeorar los síntomas del sistema nervioso. También puede causar depresión de la médula ósea, por lo que esta no puede fabricar suficientes glóbulos rojos y plaquetas. Usa la penicilamina con precaución si tienes alergia a este fármaco. También impide el funcionamiento de la vitamina B6 (piridoxina). Esto significa que necesitarás tomar dosis bajas de un suplemento B6.
- Trientina (Cuvrior, Syprine). Existe otro quelante del cobre, denominado trientina, que funciona de manera similar a la penicilamina, pero suele causar menos efectos secundarios. Sin embargo, los síntomas del sistema nervioso pueden empeorar si tomas trientina.
- Acetato de cinc (Galzin). Este medicamento evita que el cuerpo absorba el cobre de los alimentos que comes. Suele utilizarse para impedir que el cobre vuelva a acumularse luego de hacer recibido un tratamiento con penicilamina o trientina. El acetato de zinc puede utilizarse como terapia principal si no puedes tomar penicilamina ni trientina después de completar la terapia para eliminar el exceso de cobre o si no presentas síntomas. El acetato de zinc puede causarte malestar estomacal.
Tu médico también puede recomendarte diversas maneras de tratar otros síntomas de la enfermedad de Wilson.
Cirugía
Si el hígado está dañado, es posible que necesites un trasplante de hígado. Durante un trasplante de hígado, el cirujano extirpa el hígado afectado por la enfermedad y lo reemplaza con el hígado saludable de un donante.
La mayoría de los hígados trasplantados proviene de donantes fallecidos. En algunos casos, el hígado puede provenir de un donante vivo, por ejemplo, de un familiar. En ese caso, el cirujano extirpa el hígado afectado por la enfermedad y lo reemplaza con una parte del hígado del donante.
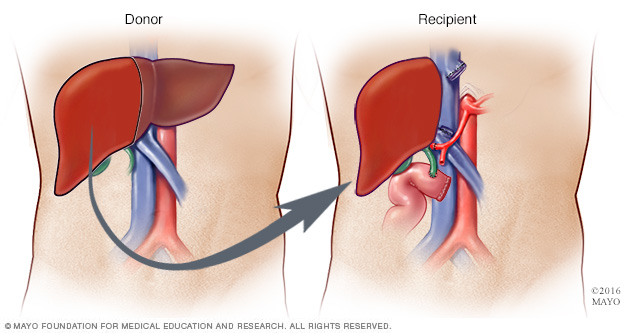
Durante la donación en vivo del hígado, los cirujanos extraen entre un 40 % y un 70 % del hígado del donante y lo colocan en el receptor.
Estilo de vida y remedios caseros
Si tienes la enfermedad de Wilson, es probable que el médico te recomiende limitar la cantidad de cobre que consumes en tu alimentación. Además, si tienes tuberías de cobre en casa, tal vez debas analizar los niveles de cobre del agua del grifo. Asegúrate de evitar los multivitamínicos que contienen cobre.
Estos son algunos alimentos que contienen mucha cantidad de cobre:
- Hígado
- Mariscos
- Hongos
- Frutos secos
- Chocolate
Pídele al equipo de atención médica más información sobre los alimentos que contienen mucha cantidad de cobre.
Preparación antes de la cita
Es probable que primero debas consultar a tu médico de familia. Luego pueden remitirte a hepatólogo, que es un médico que se especializa en hígado.
Qué puedes hacer
Cuando programes la cita médica, pregunta si debes hacer algo con anticipación, por ejemplo, cambios en tu alimentación para los análisis de sangre.
Prepara una lista de lo siguiente:
- Los síntomas y cuándo comenzaron.
- Información personal clave, como las situaciones estresantes, otras enfermedades que padezcas y cualquier antecedente familiar de la enfermedad de Wilson
- Todos los medicamentos, las vitaminas u otros suplementos que tomes, incluidas las dosis
- Preguntas para hacer al médico
Si es posible, lleva contigo a un familiar o un amigo para que te ayude a recordar la información que te dan.
Para la enfermedad de Wilson, algunas preguntas básicas para hacerle al médico son las siguientes:
- ¿Qué pruebas debo hacerme?
- ¿Qué tratamiento recomienda?
- ¿Cuáles son los efectos secundarios del tratamiento recomendado?
- ¿Hay otras opciones de tratamiento?
- Tengo estas otras enfermedades. ¿Cuál es la mejor manera de controlar estas enfermedades de manera conjunta?
- ¿Necesito limitar los tipos de alimentos que como?
- ¿Debería consultar con un especialista?
- ¿Deberían hacerle análisis a mi familia para detectar la enfermedad de Wilson?
- ¿Hay algún folleto u otro material impreso que pueda llevarme? ¿Qué sitios web me recomienda?
No dudes en hacer otras preguntas.
Qué esperar del médico
Es probable que el médico te haga varias preguntas, como las siguientes:
- ¿Están los síntomas presentes todo el tiempo o solo de vez en cuando?
- ¿Cuál es la gravedad de los síntomas?
- ¿Por cuánto tiempo has tenido estos síntomas?
- ¿Hay algo que parezca mejorar o empeorar los síntomas?
- ¿Tiene algún miembro de tu familia la enfermedad de Wilson?
Last Updated Mar 16, 2024
© 2024 Mayo Foundation for Medical Education and Research (MFMER). All rights reserved. Terms of Use