Síndrome de DiGeorge (síndrome de deleción del cromosoma 22q11.2)
Perspectiva general
El síndrome de DiGeorge, o síndrome de deleción 22q11.2, es una afección que se ocasiona cuando falta una pequeña parte del cromosoma 22. Esta deleción hace que varios sistemas corporales no se desarrollen bien.
El término síndrome de deleción 22q11.2 abarca términos que antes se consideraban afecciones diferentes. Estos términos incluyen el síndrome de DiGeorge, el síndrome velocardiofacial y otras afecciones causadas por la misma parte que falta del cromosoma 22. No obstante, las características pueden variar ligeramente.
Los problemas médicos que suelen estar relacionados con el síndrome de deleción 22q11.2 incluyen problemas cardíacos, inmunidad reducida, paladar hendido, complicaciones por niveles bajos de calcio, diversos problemas oculares y trastornos autoinmunitarios. Las complicaciones también incluyen pérdida auditiva; diferencias en el esqueleto, los riñones y los genitales, y retraso en el desarrollo con problemas conductuales y emocionales.
La cantidad y la gravedad de los síntomas relacionados con el síndrome de deleción 22q11.2 varían, pero los especialistas en diversos campos deben tratar a casi todas las personas con este síndrome.
Síntomas
Los síntomas del síndrome de DiGeorge pueden variar en función de los sistemas corporales afectados y de la gravedad de los problemas. Algunos síntomas pueden ser evidentes al nacer, pero otros pueden no aparecer hasta más tarde en la infancia o en la edad adulta.
Los síntomas del síndrome de DiGeorge pueden incluir los siguientes:
- Problemas cardíacos, como problemas en la estructura del corazón y los vasos sanguíneos, o un soplo cardíaco y cianosis, que es la piel azulada debido a la mala circulación de la sangre.
- Infecciones frecuentes.
- Rasgos faciales distintivos, como mentón poco desarrollado, orejas con aspecto diferente, ojos muy separados, párpados caídos y punta de la nariz agrandada. También puede haber facies asimétrica con el llanto. Esto ocurre cuando los músculos de un lado de la boca no se desarrollan por completo, lo que deriva en que ese lado de la boca se caiga al llorar, aunque la cara parezca equilibrada en reposo.
- Paladar hendido, que es un agujero en la parte superior de la boca, u otros problemas en el paladar.
- Dificultad para alimentarse, falta de aumento de peso o problemas estomacales.
- Pérdida auditiva.
- Poco tono muscular.
- Problemas renales.
- Visión deficiente y otros problemas oculares.
- Nivel bajo de calcio en la sangre.
- Escoliosis.
Otros síntomas pueden incluir los siguientes:
- Retraso en el crecimiento
- Retraso en el desarrollo, como retrasos para rodar, enderezarse u otros hitos del desarrollo de los bebés
- Retraso en el desarrollo del habla o voz nasal
- Retrasos en el aprendizaje o discapacidades
- Problemas de conducta
Cuándo debes consultar a un médico
Otras afecciones pueden causar síntomas como el síndrome de deleción 22q11.2. Es importante obtener el diagnóstico adecuado cuanto antes si tu hijo presenta alguno de los síntomas anteriores.
Los profesionales de atención médica pueden sospechar la existencia del síndrome de deleción 22q11.2:
- En el nacimiento. Si al nacer se observa un problema cardíaco grave, paladar hendido u otros signos típicos del síndrome de deleción 22q11.2, es probable que se realicen pruebas antes de que tu hijo salga del hospital.
- En consultas de control del niño sano. Las enfermedades o afecciones típicas del síndrome de deleción 22q11.2 pueden hacerse evidentes con el tiempo. Es posible que el profesional de atención médica de tu hijo observe algún problema durante las consultas de control del niño sano o las revisiones anuales.
Causas
Las personas tienen dos copias del cromosoma 22, una heredada del padre y otra de la madre. Si una persona tiene síndrome de DiGeorge, a una copia del cromosoma 22 le falta un segmento que consta, aproximadamente, de 30 a 40 genes. Muchos de estos genes aún no se han identificado de manera concreta ni se comprenden. La región del cromosoma 22 que se elimina se conoce como 22q11.2.
La deleción de los genes del cromosoma 22, por lo general, se produce como un evento aleatorio en los espermatozoides del padre o en el óvulo de la madre. También puede producirse en las primeras etapas del desarrollo fetal. En casos pocos frecuentes, la deleción se transmite a un hijo del padre o la madre que también tiene una deleción en el cromosoma 22, pero que tener puede menos síntomas o síntomas leves.
Complicaciones
Las partes del cromosoma 22 que faltan en el síndrome de DiGeorge afectan el desarrollo de varios sistemas corporales. En consecuencia, la afección puede causar varios errores durante el desarrollo fetal.
- Problemas cardíacos. El síndrome de deleción 22q11.2 suele causar problemas cardíacos que pueden derivar en que la sangre rica en oxígeno sea demasiado escasa. Por ejemplo, entre los problemas puede incluirse un agujero entre las dos cámaras inferiores del corazón, conocido como defecto del tabique ventricular. También puede haber un tronco arterioso, que es solo un vaso grande en lugar de dos vasos que salen del corazón. O bien puede haber una tetralogía de Fallot, cuatro problemas en la estructura del corazón.
- Hipoparatiroidismo. Las cuatro glándulas paratiroides del cuello regulan los niveles de calcio y de fósforo en el cuerpo. El síndrome de deleción 22q11.2 puede hacer que las glándulas paratiroides sean más pequeñas de lo normal y produzcan muy poca hormona paratiroidea. Esto deriva en hipoparatiroidismo. Esta afección hace que en la sangre haya niveles bajos de calcio y niveles altos de fósforo.
- Disfunción del timo. El timo, glándula que se encuentra debajo del esternón, es donde maduran las células T, un tipo de glóbulo blanco. Las células T maduras ayudan a combatir infecciones. En los niños con síndrome de deleción 22q11.2, el timo puede ser pequeño o puede no estar. Esto lleva a una función inmunitaria deficiente e infecciones frecuentes y graves.
- Paladar hendido. Una afección común del síndrome de deleción 22q11.2 es el paladar hendido, un agujero en la parte superior de la boca, con labio hendido o sin este. Otros problemas menos visibles en la estructura del paladar pueden dificultar la acción de tragar o la emisión de determinados sonidos del habla.
- Rasgos faciales distintivos. En algunas personas con síndrome de deleción 22q11.2, se pueden presentar diversos rasgos faciales particulares. Entre ellos, se incluyen orejas pequeñas y de posición baja; aberturas oculares poco anchas, fisuras palpebrales; párpados caídos; cara relativamente larga; punta de la nariz agrandada, bulbosa; o surco corto o aplanado en el labio superior.
- Problemas de aprendizaje, de comportamiento y de salud mental. El síndrome de deleción 22q11.2 puede causar problemas en el desarrollo y la actividad del cerebro, lo que causa problemas de aprendizaje, para socializar, del desarrollo o de conducta. Son comunes los retrasos en el desarrollo del habla en los niños pequeños, así como la dificultad para aprender. Algunos niños desarrollan trastorno por déficit de atención e hiperactividad o trastorno del espectro autista. Más adelante, hay un mayor riesgo de depresión, ansiedad y otras afecciones de salud mental.
- Afecciones autoinmunes. Las personas con síndrome de deleción 22q11.2 también pueden tener mayor riesgo de padecer afecciones autoinmunes, como artritis reumatoide o enfermedad de Graves.
- Otros problemas. Muchas enfermedades pueden estar relacionadas con el síndrome de deleción 22q11.2, como problemas de audición, problemas oculares y una función renal deficiente.
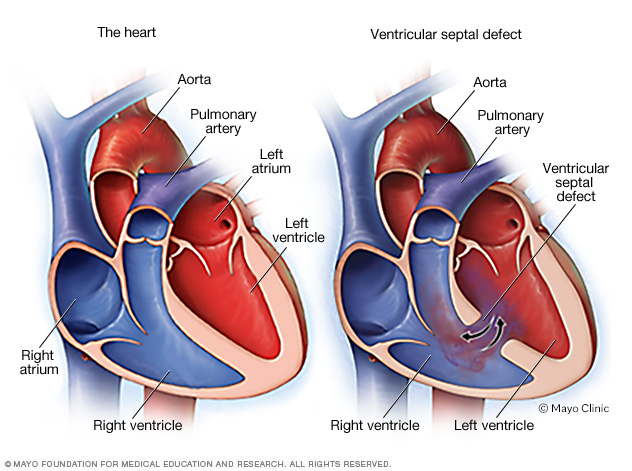
El defecto del tabique ventricular consiste en un orificio en el corazón que está presente desde el nacimiento (defecto cardíaco congénito). El orificio se encuentra entre las cavidades inferiores del corazón (ventrículos derecho e izquierdo). Hace que la sangre oxigenada regrese a los pulmones en lugar de bombearse al resto del cuerpo.

En el tronco arterioso, un vaso sanguíneo grande sale del corazón, en vez de dos vasos sanguíneos separados. Además, suele observarse un orificio en la pared que se encuentra entre las cavidades cardíacas inferiores (ventrículos). Este orificio se conoce como comunicación auriculoventricular. En el tronco arterioso, la sangre oxigenada (que se muestra en rojo) y la sangre con poco oxígeno (que se muestra en azul) se mezclan. La sangre mezclada se muestra en púrpura. Esta sangre no tiene suficiente oxígeno para satisfacer las necesidades del cuerpo.
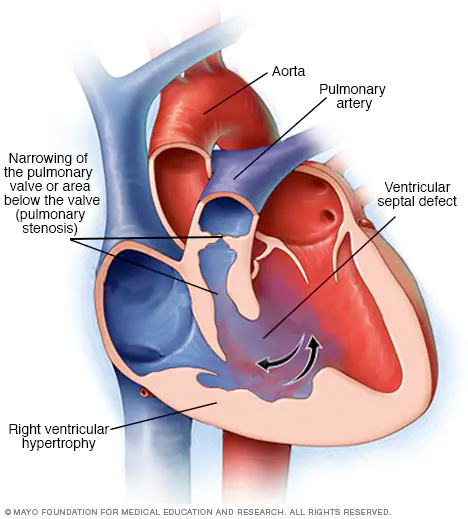
La tetralogía de Fallot es una combinación de cuatro defectos cardíacos presentes al nacer. Hay un orificio en el corazón denominado defecto septal ventricular. También hay un estrechamiento de la válvula pulmonar u otra zona a lo largo del conducto que une el corazón con los pulmones. El estrechamiento de la válvula pulmonar se conoce como estenosis pulmonar. La arteria principal del cuerpo, llamada aorta, se encuentra desplazada. La pared de la cavidad inferior derecha del corazón está engrosada, lo que se denomina hipertrofia del ventrículo derecho. La tetralogía de Fallot cambia la manera en la que la sangre fluye a través del corazón y hacia el resto del cuerpo.
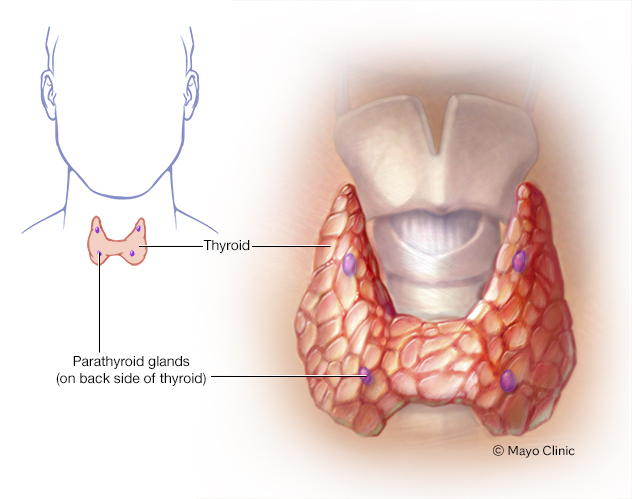
Las cuatro pequeñas glándulas paratiroides, que están cerca de la tiroides, liberan la hormona paratiroidea. La hormona participa en el control de los niveles de los minerales calcio y fósforo en el cuerpo.
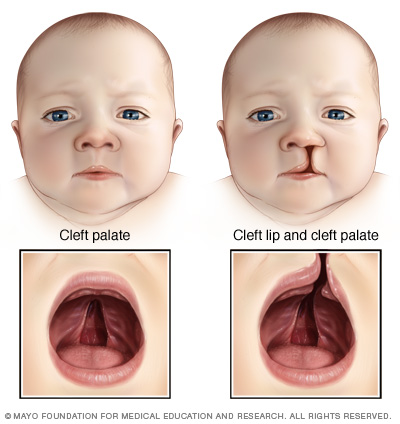
Una hendidura del paladar es una abertura o una división en el techo de la boca que ocurre cuando el tejido no se fusiona durante el desarrollo en el útero. La hendidura del paladar suele comprender una división (hendidura) en el labio superior (labio leporino), pero puede ocurrir sin que el labio se vea afectado.
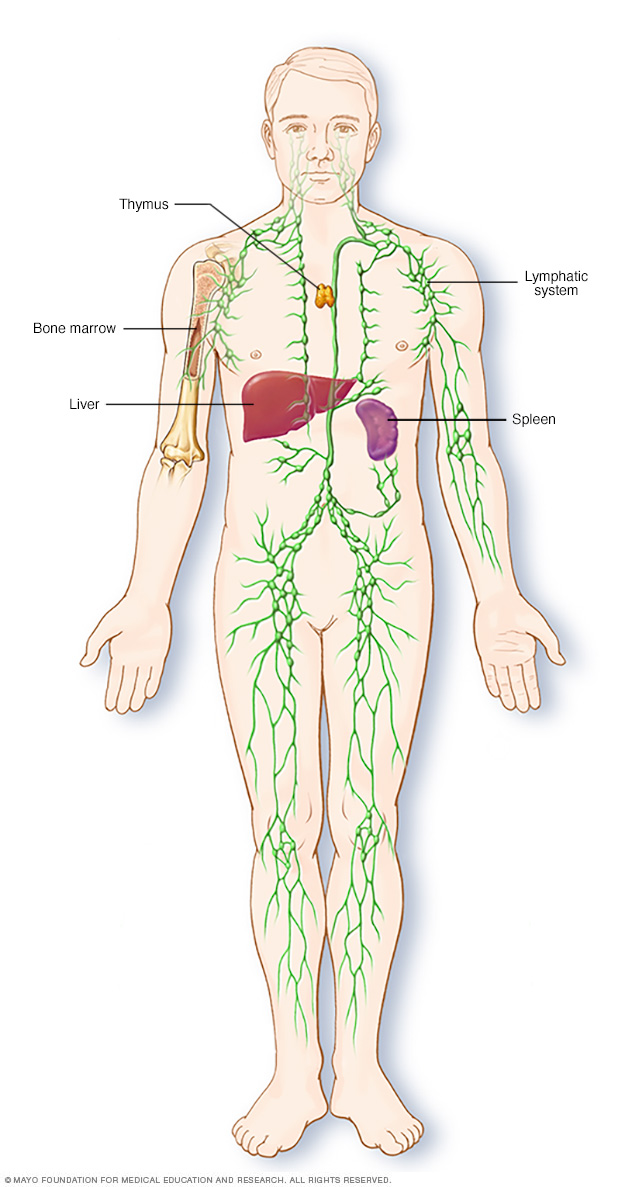
El sistema linfático forma parte del sistema inmunitario del cuerpo, que protege de infecciones y enfermedades. El sistema linfático incluye el bazo, el timo, los ganglios y los canales linfáticos, así como también las amígdalas y las adenoides.
Prevención
En algunos casos, el padre o la madre afectado puede transmitir el síndrome DiGeorge a un hijo. Si te preocupan los antecedentes familiares del síndrome de deleción 22q11.2 o si ya tienes un hijo con este síndrome, puedes acudir a un médico especializado en afecciones genéticas. A este médico se lo conoce como genetista. También puedes acudir a un consejero genético para que te ayude a planificar los próximos embarazos.
Diagnóstico
Un diagnóstico de síndrome de DiGeorge, o síndrome de deleción 22q11.2, se basa, principalmente, en un análisis de laboratorio que puede detectar la deleción en el cromosoma 22. Es probable que el profesional de atención médica de tu hijo solicite esta prueba si tu hijo presenta lo siguiente:
- Una combinación de problemas médicos o afecciones que sugieren el síndrome de deleción 22q11.2.
- Un problema cardíaco, ya que determinados problemas cardíacos suelen estar relacionados con el síndrome de deleción 22q11.2.
En algunos casos, un niño puede tener una combinación de afecciones que sugieren el síndrome de deleción 22q11.2, pero el análisis de laboratorio no indica que falta una parte en el cromosoma 22.
Tratamiento
Aunque el síndrome de DiGeorge, o síndrome de deleción 22q11.2, no tiene cura, los tratamientos suelen corregir problemas importantes, como problemas cardíacos o paladar hendido. Otros problemas de salud y problemas de desarrollo, salud mental y de comportamiento pueden abordarse o controlarse según sea necesario.
Los tratamientos y las terapias contra el síndrome de deleción 22q11.2 pueden comprender tratamientos para lo siguiente:
- Hipoparatiroidismo. Tomar suplementos de calcio y vitamina D según las indicaciones del profesional de atención médica suele ayudar a controlar el hipoparatiroidismo. En algunos casos, es posible que te recomienden otros suplementos.
- Problemas cardíacos. La mayoría de los problemas cardíacos relacionados con el síndrome de deleción 22q11.2 requieren cirugía poco después del nacimiento para reparar el corazón y mejorar el suministro de sangre rica en oxígeno.
- Funcionamiento limitado del timo. Si tu hijo tiene cierta limitación en el funcionamiento del timo, las infecciones pueden ser frecuentes, pero no necesariamente graves. Estas infecciones, a menudo resfriados e infecciones en el oído, por lo general, se tratan como se haría en cualquier niño. La mayoría de los niños con una actividad limitada del timo sigue un cronograma habitual de vacunas. La función del sistema inmunitario mejora con la edad en la mayoría de los niños que tienen un daño moderado en el timo.
- Disfunción grave del timo. Si el daño del timo es grave o si tu hijo no tiene el timo, corre el riesgo de sufrir diversas infecciones graves. El tratamiento puede requerir un trasplante de tejido del timo y de células especializadas de la médula ósea o de células sanguíneas especializadas que combaten las enfermedades.
- Paladar hendido. Un paladar hendido u otras características inusuales del paladar y el labio suelen poder repararse mediante cirugía.
- Desarrollo general. Es probable que tu hijo se beneficie de una variedad de terapias; entre ellas, la terapia del habla, la terapia ocupacional y la terapia del desarrollo. En los Estados Unidos, los programas de intervención temprana que brindan estos tipos de terapia, por lo general, están disponibles a través del departamento de salud estatal o del condado.
- Cuidado de la salud mental. Se puede recomendar tratamiento si a tu hijo se le diagnostica posteriormente trastorno por déficit de atención e hiperactividad, trastorno del espectro autista, depresión, u otros tipos de afecciones de salud mental o del comportamiento.
- Manejo de otras afecciones. Estas afecciones pueden incluir problemas de alimentación y crecimiento, problemas de audición o visuales y otras enfermedades.
Equipo de atención médica
Dado que el síndrome de deleción 22q11.2 puede derivar en muchos problemas, es posible que varios especialistas ayuden a diagnosticar afecciones específicas, recomienden tratamientos y proporcionen cuidados. Este equipo cambiará a medida que cambien las necesidades de tu hijo.
Los especialistas del equipo de atención médica de tu hijo pueden incluir a estos profesionales y a otros, según sea necesario:
- Pediatra, especialista en salud infantil
- Genetista, experto en afecciones hereditarias
- Cardiólogo, especialista en corazón
- Inmunólogo, especialista en el sistema inmunitario
- Otorrinolaringólogo, especialista en oído, nariz y garganta
- Especialista en enfermedades infecciosas
- Endocrinólogo, especialista en afecciones hormonales
- Cirujano especialista en oído, nariz y garganta, especializado en la corrección de trastornos tales como paladar hendido
- Cirujano cardiovascular, especializado en corregir problemas cardíacos
- Terapeuta ocupacional para desarrollar habilidades prácticas y cotidianas
- Terapeuta del habla para mejorar la capacidad de hablar
- Terapeuta del desarrollo para trabajar en comportamientos y habilidades sociales apropiados para la edad
- Profesional de la salud mental, como un psicólogo o un psiquiatra pediátrico
Estrategias de afrontamiento, y apoyo
Tener un hijo con el síndrome de DiGeorge, o síndrome de deleción 22q11.2, es desafiante. Puedes tener que enfrentarte a múltiples problemas de salud y tratamientos. Para ayudarte a satisfacer las necesidades de tu hijo y las tuyas propias, pregúntale al equipo de atención médica sobre las organizaciones que ofrecen material educativo, grupos de apoyo y otros recursos para padres de niños con síndrome de deleción 22q11.2.
Preparación antes de la cita
El médico de tu hijo u otro profesional de atención médica puede sospechar que el síndrome DiGeorge está presente en el nacimiento. Si ese es el caso, es probable que se realicen pruebas y tratamientos antes de que tu hijo salga del hospital.
El profesional de atención médica de tu hijo buscará problemas de desarrollo en las revisiones periódicas y te comentará sobre cualquier inquietud. Es importante que lleves a tu hijo a todas las consultas de control del niño sano y las citas médicas anuales programadas con regularidad.
A continuación, encontrarás información que te ayudará a prepararte para la cita de tu hijo.
Qué puedes hacer
Si el profesional de atención médica de familia o pediatra cree que tu hijo presenta signos del síndrome de deleción 22q11.2, las preguntas básicas que debes hacer son las siguientes:
- ¿Qué pruebas se necesitarán?
- ¿Cuándo obtendremos los resultados de las pruebas?
- ¿A qué especialistas remitirán a mi hijo?
- ¿Qué enfermedades relacionadas con este síndrome necesitan tratarse de inmediato? ¿Qué enfermedad es más importante?
- ¿Cómo me ayudará a vigilar los problemas de salud y desarrollo de mi hijo?
- ¿Me podría recomendar materiales educativos y servicios de apoyo locales sobre este síndrome?
- ¿Qué servicios están disponibles para el desarrollo de la primera infancia?
Qué esperar del médico
Prepárate para responder las preguntas que el profesional de atención médica puede hacerte, como las siguientes:
- ¿Tiene tu bebé algún problema para alimentarse?
- ¿Se ve tu bebé desanimado, débil o enfermo?
- ¿Está tu hijo alcanzando determinados hitos del desarrollo, como rodar, empujar para levantarse, sentarse, gatear, caminar o hablar?
- ¿Ves algún comportamiento que te preocupe?
Estar listo para las preguntas te ayudará a aprovechar al máximo el tiempo con el profesional de atención médica de tu hijo.
Last Updated Apr 20, 2024
© 2024 Mayo Foundation for Medical Education and Research (MFMER). All rights reserved. Terms of Use