Síndrome de Marfan
Perspectiva general
El síndrome de Marfan es un trastorno hereditario que afecta el tejido conectivo, es decir, las fibras que sostienen y sujetan los órganos y otras estructuras del cuerpo. El síndrome de Marfan afecta más frecuentemente el corazón, los ojos, los vasos sanguíneos y el esqueleto.
Las personas con síndrome de Marfan generalmente son altas y delgadas, y sus brazos, piernas, dedos de los pies y las manos son inusualmente largos. El daño causado por el síndrome de Marfan puede ser leve o grave. Si la aorta (el vaso sanguíneo de gran tamaño que lleva sangre desde el corazón hacia el resto del cuerpo) se ve afectada, la afección puede poner en riesgo la vida.
El tratamiento en general comprende medicamentos para mantener una presión arterial baja y así reducir la tensión sobre la aorta. Es vital realizar controles regulares para verificar la progresión del daño. Muchas personas con síndrome de Marfan con el tiempo requieren cirugía preventiva para reparar la aorta.
Síntomas
Los signos y síntomas del síndrome de Marfan varían ampliamente, aun entre miembros de la misma familia, ya que el trastorno puede afectar a muchas zonas distintas del cuerpo. Algunas personas experimentan solo efectos leves, pero otras contraen complicaciones que ponen en riesgo la vida.
Las características del síndrome de Marfan pueden ser:
- Una contextura alta y delgada
- Brazos, piernas y dedos desproporcionadamente largos
- Esternón que sobresale o se hunde
- Paladar alto y arqueado, y dientes apiñados
- Soplos cardíacos
- Miopía extrema
- Espina dorsal anormalmente curvada
- Pie plano
Cuándo debes consultar con un médico
Si crees que tú o tu hijo pueden tener el síndrome de Marfan, habla con tu médico o pediatra. Si tu médico sospecha que tienes un problema, es posible que te remita a un especialista para una evaluación más profunda.
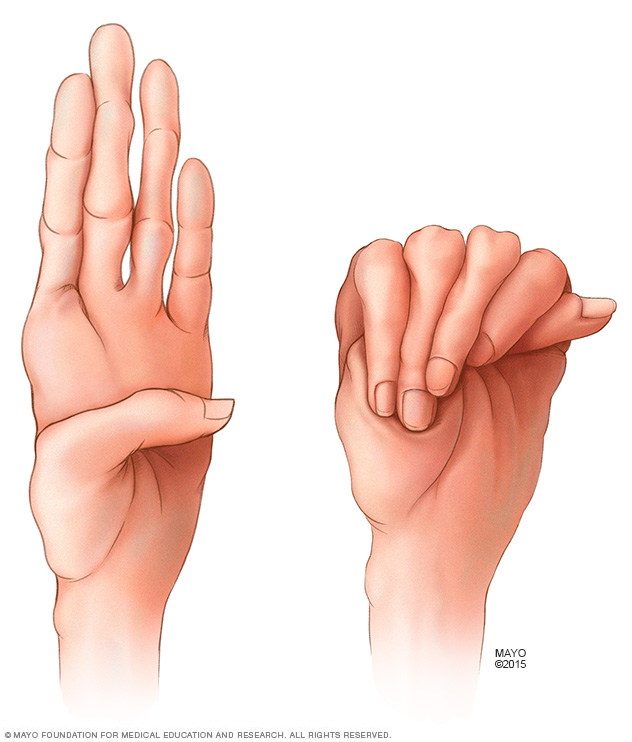
Es típico que las personas que tienen el síndrome de Marfan tengan dedos especialmente largos. Es normal que los pulgares les sobresalgan de la mano cuando cierran el puño.

El síndrome de Marfan es un trastorno genético que hace que las personas tengan brazos, piernas y dedos inusualmente largos. Es posible que el médico quiera medirte la distancia de ambos brazos extendidos si cree que puedes padecer este trastorno.
Causas
El síndrome de Marfan es provocado por un defecto en el gen que le permite al cuerpo producir una proteína que ayuda a darle elasticidad y fuerza al tejido conectivo.
La mayoría de las personas con síndrome de Marfan hereda el gen anormal de uno de los padres con el trastorno. Cada hijo de un progenitor afectado tiene una probabilidad de 50/50 de heredar el gen defectuoso. En alrededor del 25 % de las personas con síndrome de Marfan, el gen anormal no proviene de ninguno de sus padres. En estos casos, se desarrolla una nueva mutación de forma espontánea.
Factores de riesgo
El síndrome de Marfan afecta a los hombres y a las mujeres de igual forma, y se presenta en todas las razas y grupos étnicos. Debido a que es una enfermedad genética, el mayor factor de riesgo del síndrome de Marfan es que uno de los padres tenga este trastorno.
Complicaciones
Como el síndrome de Marfan puede afectar casi cualquier parte del cuerpo, es posible que provoque una gran variedad de complicaciones.
Complicaciones cardiovasculares
Las complicaciones más peligrosas del síndrome de Marfan afectan el corazón y los vasos sanguíneos. El tejido conjuntivo defectuoso puede debilitar la aorta, la arteria grande que surge desde el corazón y suministra la sangre al organismo.
- Aneurisma de la aorta. La presión de la sangre que sale del corazón puede hacer que se forme una protuberancia en la pared de la aorta, como un punto débil en un neumático. En las personas con síndrome de Marfan, es más probable que esto ocurra en la raíz aórtica, donde la arteria sale del corazón.
- Disección aórtica. La pared de la aorta se compone de capas. La disección se produce cuando un desgarro pequeño en la capa más interna de la pared de la aorta permite que la sangre se filtre entre las capas internas y externas de la pared. Esto puede provocar dolor intenso en el pecho o en la espalda. Una disección aórtica debilita la estructura de los vasos sanguíneos y puede provocar una rotura, que posiblemente sea mortal.
- Malformaciones de las válvulas. En personas que tienen el síndrome de Marfan, el tejido de las válvulas cardíacas puede ser débil. Esto puede provocar el estiramiento del tejido valvular y un funcionamiento anormal de las válvulas. Cuando las válvulas cardíacas no funcionan correctamente, el corazón a menudo tiene que trabajar más para compensar. Esto puede finalmente provocar una insuficiencia cardíaca.
Complicaciones oculares
Las complicaciones en los ojos pueden comprender las siguientes:
- Luxación del cristalino. La lente de enfoque dentro del ojo puede moverse del lugar si se debilitan sus estructuras de soporte. El término médico para este problema es "ectopia del cristalino", y este ocurre en más de la mitad de las personas que tienen síndrome de Marfan.
- Problemas de retina. El síndrome de Marfan también aumenta el riesgo de un desprendimiento o desgarro de la retina, el tejido sensible a la luz que recubre la pared posterior del ojo.
- Aparición temprana de glaucoma o cataratas. Las personas que tienen síndrome de Marfan suelen desarrollar estos problemas en los ojos a una edad más temprana. El glaucoma provoca que la presión intraocular aumente, lo que puede dañar el nervio óptico. Las cataratas son partes opacas en el cristalino del ojo, que normalmente es transparente.
Complicaciones óseas
El síndrome de Marfan aumenta el riesgo de curvas anormales en la columna vertebral, como la escoliosis. También puede afectar el desarrollo normal de las costillas, ya que puede provocar que el esternón sobresalga o se hunda en el pecho. El dolor en los pies y en la espalda es frecuente con el síndrome de Marfan.
Complicaciones del embarazo
El síndrome de Marfan puede debilitar las paredes de la aorta, la arteria principal que sale del corazón. Durante el embarazo, el corazón bombea más sangre que lo habitual. Esto puede generar más presión en la aorta, lo que aumenta el riesgo de ruptura o disección mortal.
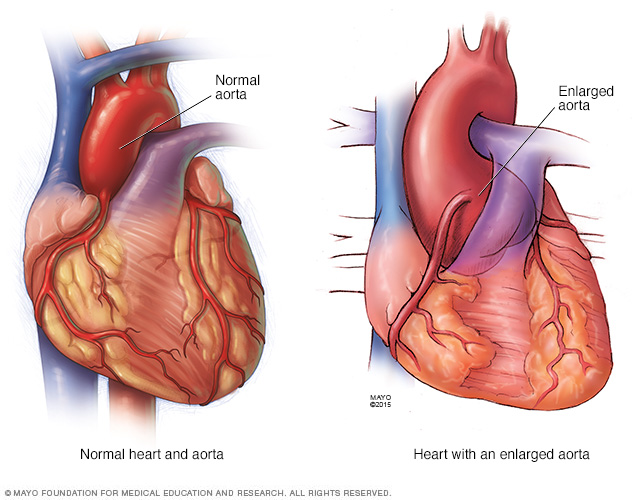
La presión de la sangre que sale del corazón puede hacer que se forme un bulto en la pared de la aorta, como un punto débil en un neumático. En las personas que tienen el síndrome de Marfan, lo más probable es que esto ocurra en la raíz aórtica, donde la arteria sale del corazón.
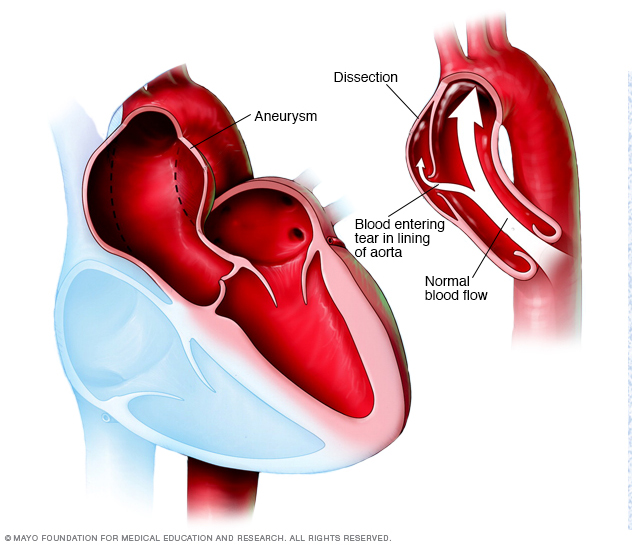
Un aneurisma aórtico se produce cuando un punto débil en la pared de la aorta comienza a abultarse (izquierda). Esto puede ocurrir en cualquier parte de la aorta. Padecer un aneurisma aumenta el riesgo de un desgarro en el revestimiento de la aorta, como se muestra en la imagen a la derecha.
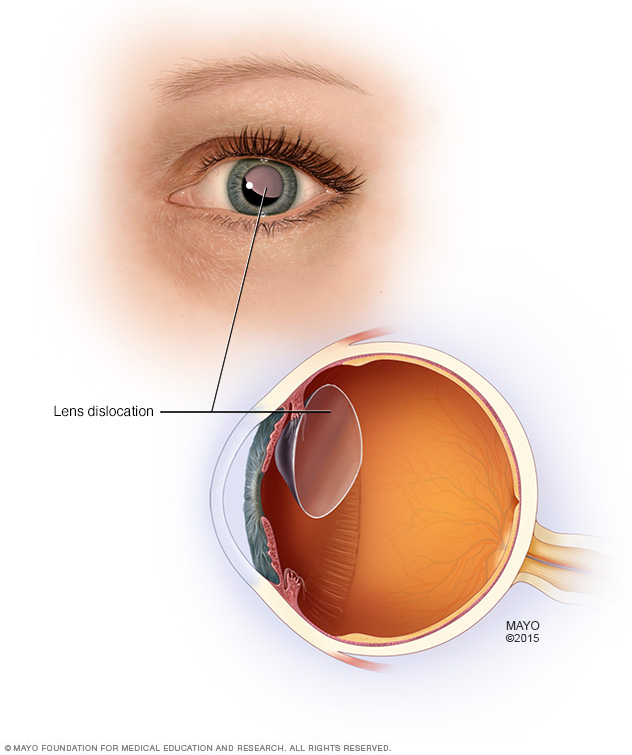
Algunas personas con síndrome de Marfan pueden sufrir la luxación del cristalino del ojo.
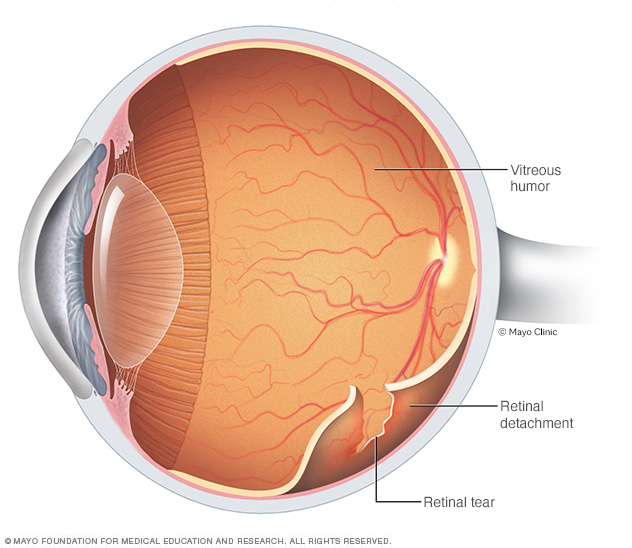
El desprendimiento de retina es una situación de emergencia en la que una fina capa de tejido (la retina) en la parte posterior del ojo se desprende de la capa de vasos sanguíneos que le proporciona oxígeno y nutrientes. El desprendimiento de retina suele estar acompañado de destellos de luz y cuerpos flotantes en la visión.
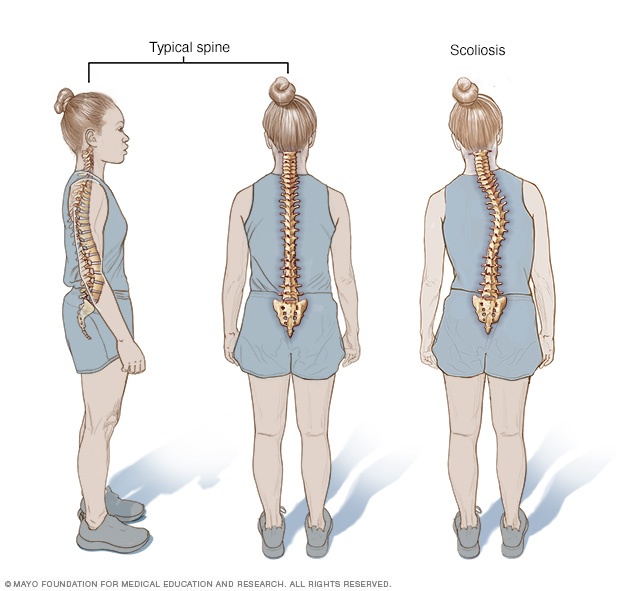
Desde una vista lateral, la forma de la columna vertebral normal es de una S alargada: la parte superior de la espalda se arquea hacia afuera y la región lumbar se curva levemente hacia adentro. Sin embargo, desde una vista trasera, la columna vertebral parece una línea recta desde la base del cuello hasta el coxis. La escoliosis es una desviación lateral de la columna vertebral.
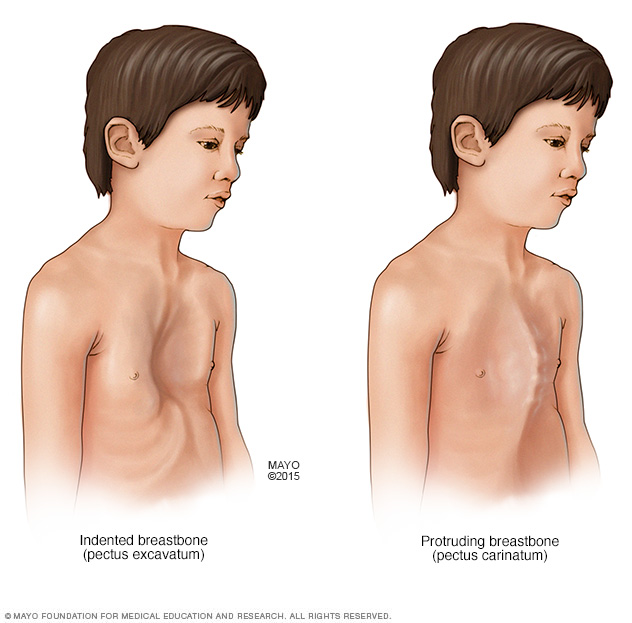
El síndrome de Marfan puede interferir en el desarrollo normal de las costillas, lo que puede hacer que el esternón sobresalga o parezca hundido en el tórax.
Diagnóstico
Para los médicos, puede ser difícil diagnosticar el síndrome de Marfan, ya que muchos trastornos del tejido conjuntivo presentan signos y síntomas similares. Incluso entre los miembros de la misma familia, los signos y síntomas del síndrome de Marfan varían ampliamente, tanto en lo que respecta a las características como en lo que respecta a la gravedad.
Deben estar presentes ciertas combinaciones de los síntomas y de los antecedentes familiares para confirmar el diagnóstico del síndrome de Marfan. En algunos casos, una persona puede tener ciertas características del síndrome de Marfan, pero no las suficientes para diagnosticar el trastorno.
Estudios cardíacos
Si el médico sospecha que puedes tener síndrome de Marfan, una de las primeras pruebas que puede recomendar es un ecocardiograma. Esta prueba utiliza ondas sonoras para capturar imágenes en tiempo real del corazón en movimiento. Controla el estado de las válvulas cardíacas y el tamaño de la aorta. Otras opciones de imágenes del corazón comprenden la tomografía computarizada (TC) o la resonancia magnética (RM).
Si te diagnostican síndrome de Marfan, vas a tener que hacerte estudios de diagnóstico por imágenes en forma regular para controlar el tamaño y el estado de la aorta.
Exámenes oculares
Exámenes de la vista que pueden ser necesarios, entre ellos:
- Examen con lámpara de hendidura. Esta prueba detecta si hay luxación del cristalino, cataratas o desprendimiento de retina. Para este examen, los ojos se tendrán que dilatar completamente con gotas.
- Prueba de presión ocular. Para verificar si hay glaucoma, el oftalmólogo puede medir la presión dentro del globo ocular tocándolo con una herramienta especial. Generalmente, se utilizan gotas anestésicas para los ojos antes de esta prueba.
Análisis genéticos
Las pruebas genéticas a menudo se usan para confirmar el diagnóstico del síndrome de Marfan. Si se encuentra una mutación de Marfan, se puede examinar a los miembros de la familia para determinar si también están afectados. Posiblemente desees hablar con un consejero genético antes de comenzar a formar una familia para saber cuáles son las probabilidades de trasmitir el síndrome de Marfan a tus futuros hijos.
Tratamiento
Si bien no existe una cura para el síndrome de Marfan, el tratamiento se centra en la prevención de las diversas complicaciones de esta enfermedad. Para lograr esto, tendrás que controlarte regularmente para detectar si los daños provocados por la enfermedad avanzan.
En el pasado, las personas que tenían el síndrome de Marfan a menudo morían jóvenes. Con controles periódicos y un tratamiento moderno, la mayoría de las personas con síndrome de Marfan ahora puede esperar llevar una vida más normal.
Medicamentos
Los médicos a menudo recetan medicamentos que reducen la presión arterial para ayudar a prevenir la dilatación de la aorta y para reducir el riesgo de disección y rotura.
Terapia
En general, los problemas de visión asociados con una lente dislocada en el ojo se pueden corregir con anteojos o lentes de contacto.
Cirugías y otros procedimientos
Según los signos y síntomas, los procedimientos pueden comprender los siguientes:
- Reparación aórtica. Si el diámetro de la aorta alcanza aproximadamente 2 pulgadas (50 milímetros) o si se agranda rápidamente, el médico puede recomendar hacer una operación para reemplazar una porción de la aorta por un tubo de material sintético. Esto puede ayudar a prevenir una ruptura potencialmente mortal. Quizá también sea necesario reemplazar la válvula aórtica.
- Tratamientos para la escoliosis. Cuando hay escoliosis significativa, es necesaria una consulta con un experto en columna vertebral. En algunos casos, se necesitan aparatos ortopédicos y cirugía.
- Corrección del esternón. Existen opciones quirúrgicas para corregir el aspecto de un esternón hundido o sobresaliente. Como a menudo se considera que estas operaciones son para fines estéticos, es posible que tu seguro no cubra los costos.
- Cirugía ocular. Si se han desgarrado partes de la retina o si se han desprendido de la parte posterior del ojo, generalmente, la reparación quirúrgica da buenos resultados. Si tienes cataratas, el cristalino opacado puede reemplazarse con un cristalino artificial.
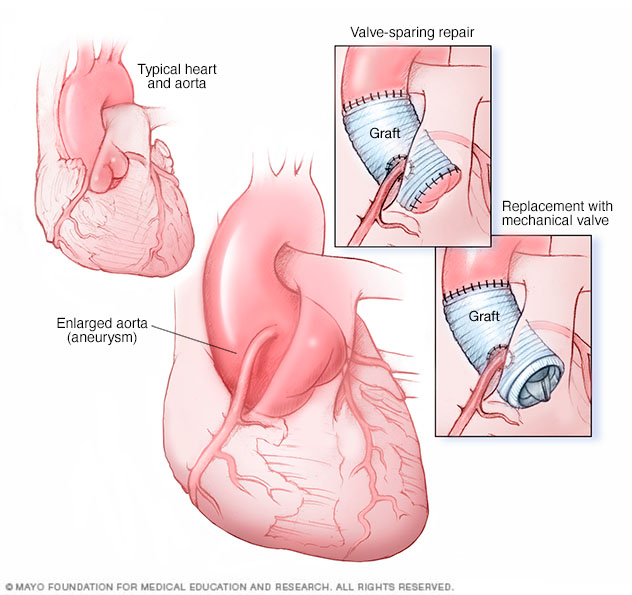
Un procedimiento de aneurisma de la raíz aórtica ascendente se puede hacer de dos maneras. La reparación de raíz aórtica con preservación de válvula (imagen superior derecha) reemplaza el segmento agrandado de la aorta con una cánula artificial que se conoce como injerto. La válvula aórtica queda en su lugar. En el reemplazo de la válvula aórtica y la raíz aórtica (imagen inferior derecha), se retiran la válvula aórtica y parte de la aorta. Se reemplaza el segmento de la aorta con un injerto. Una válvula biológica o mecánica reemplaza la válvula.
Estilo de vida y remedios caseros
Es posible que tengas que evitar los deportes competitivos y ciertas actividades recreacionales si tienes un mayor riesgo de disección o rotura aórtica. El aumento de la presión arterial, frecuente en actividades como levantar peso, genera tensión extra en la aorta. Actividades menos intensas, como una caminata a paso ligero, jugar a los bolos, dobles de tenis o golf, son en general más seguras.
Estrategias de afrontamiento, y apoyo
Vivir con un trastorno genético puede ser extremadamente difícil tanto para niños como para adultos. Los adultos pueden preguntarse cómo afectará la enfermedad a su profesión, sus relaciones y su autoestima. Y probablemente les preocupe transmitir el gen defectuoso a sus hijos.
Pero el síndrome de Marfan puede ser aún más difícil en las personas jóvenes, sobre todo porque la timidez propia de la infancia y de la adolescencia se puede exacerbar por el efecto de la enfermedad sobre la apariencia, el desempeño académico y las habilidades motoras.
Cómo ayudar a los niños a enfrentar situaciones difíciles
Trabajar en conjunto––los padres, los profesores y los profesionales médicos––puede brindar a los niños apoyo emocional y soluciones prácticas para algunos de los aspectos más preocupantes de la enfermedad. Por ejemplo, los niños con síndrome de Marfan pueden tener dificultades en la escuela debido a problemas de visión que pueden corregirse con anteojos o lentes de contacto.
Para la mayoría de las personas jóvenes, las preocupaciones estéticas son al menos tan importantes como las académicas. Los padres pueden ayudar cuando anticipan estos problemas y ofrecen soluciones, como las siguientes:
- Lentes de contacto en lugar de anteojos
- Un dispositivo de inmovilización para la escoliosis
- Procedimientos dentales para los dientes apiñados
- Ropa que favorezca una contextura delgada y alta
Grupos de apoyo
A quienes tienen síndrome de Marfan les es útil conversar con otras personas que se enfrentan a desafíos similares. La Fundación Marfan brinda diversos servicios de apoyo en línea.
Preparación antes de la cita
El síndrome de Marfan puede afectar muchas partes diferentes del cuerpo, por lo que posiblemente tengas que consultar a una gran variedad de médicos especialistas, como los siguientes:
- Un cardiólogo, un médico que se especializa en tratar los trastornos cardíacos y de los vasos sanguíneos
- Un oftalmólogo, un médico que se especializa en tratar los trastornos oculares
- Un ortopedista, un médico que se especializa en tratar los problemas estructurales del esqueleto
- Un genetista, un médico que se especializa en tratar los trastornos genéticos
Para aprovechar mejor el tiempo de la consulta, planifica con anterioridad y ten disponible la información importante, como la siguiente:
- Descripciones detalladas de todos tus síntomas
- Detalles de tus antecedentes médicos pasados, incluida cualquier cirugía anterior
- Informes de radiografías y ecocardiogramas pasados, que a menudo pueden enviarse de manera electrónica
- Una lista de todos los medicamentos y suplementos que tomes
Qué esperar del médico
Los médicos querrán saber cuáles son tus síntomas específicos y si alguien en tu familia tenía el síndrome de Marfan o experimentó alguna discapacidad o muerte tempranas relacionadas con el corazón y sin causa aparente.
Last Updated Mar 22, 2024
© 2024 Mayo Foundation for Medical Education and Research (MFMER). All rights reserved. Terms of Use