Congenital adrenal hyperplasia
Overview
Congenital adrenal hyperplasia (CAH) is the medical name for a group of genetic conditions that affect the adrenal glands. The adrenal glands are a pair of walnut-sized organs above the kidneys. They make important hormones, including:
- Cortisol. This controls the body's response to illness or stress.
- Mineralocorticoids such as aldosterone. These control sodium and potassium levels.
- Androgens such as testosterone. These sex hormones are needed for growth and development in both males and females.
In people with CAH, a gene change results in a lack of one of the enzyme proteins needed to make these hormones.
The two major types of congenital adrenal hyperplasia are:
- Classic CAH. This type is rarer and more serious. It's usually found by tests at birth or in early infancy.
- Nonclassic CAH. This type is milder and more common. It may not be found until childhood or early adulthood.
There is no cure for congenital adrenal hyperplasia. But with proper treatment, most people who have CAH can lead full lives.
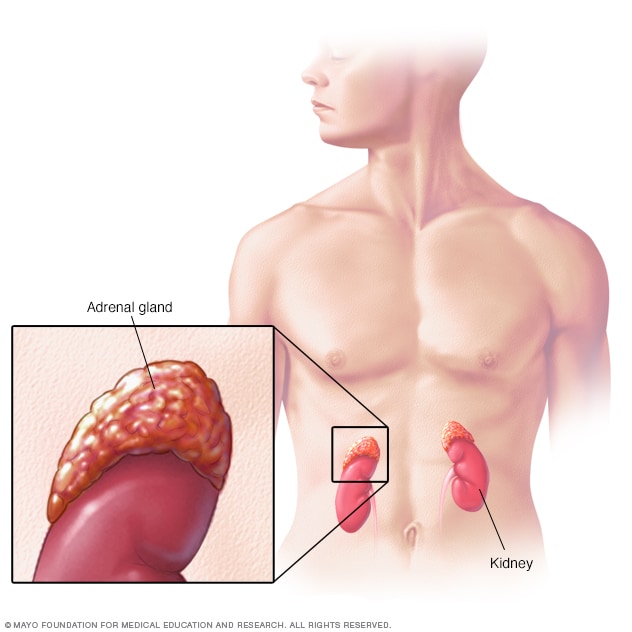
Located on top of each kidney, the adrenal glands make hormones that help regulate metabolism, the immune system, blood pressure and other important functions. Although small, these glands control much of what happens in the body.
Symptoms
Symptoms of CAH vary. The symptoms depend on which gene is affected. They also depend on how greatly the adrenal glands lack one of the enzymes needed to make hormones. With CAH, the hormones that the body needs to work properly are thrown out of balance. That may lead to too little cortisol, too little aldosterone, too many androgens or a mix of these issues.
Classic CAH
Symptoms of classic CAH can include:
- Not enough cortisol. With classic CAH, the body doesn't make enough of the hormone cortisol. This can cause problems keeping blood pressure, blood sugar and energy at healthy levels. It also can cause problems during physical stress such as illness.
- Adrenal crisis. People with classic CAH can be seriously affected by a lack of cortisol, aldosterone or both. This is known as an adrenal crisis. It can be life-threatening.
-
External genitals that don't look typical. In female infants, some parts of the genitals on the outside of the body may look different than usual. For instance, the clitoris may be enlarged and resemble a penis. The labia may be partly closed and look like a scrotum. The tube through which urine leaves the body and the vagina may be one opening instead of two separate openings. The uterus, fallopian tubes and ovaries often develop in a typical manner.
Male infants with CAH often have genitals that look typical but sometimes are enlarged.
-
Too much androgen. An excess of the male sex hormone androgen can lead to short height and early puberty for children. Pubic hair and other signs of puberty may appear at a very early age. Serious acne also may occur.
Extra androgen hormones in females may lead to facial hair, more body hair than usual and a deeper voice.
- Altered growth. Children may grow fast. And their bones could be more developed than is typical for their age. Final height may be shorter than average.
- Fertility issues. These can include irregular menstrual periods or not having periods at all. Some women with classic CAH may have trouble becoming pregnant. Fertility issues sometimes can occur in men.
Nonclassic CAH
Often, there are no symptoms of nonclassic CAH when a baby is born. Some people with nonclassic CAH never have symptoms. The condition is not found on routine infant blood screening tests. If symptoms occur, they usually appear in late childhood or early adulthood.
Females who have nonclassic CAH may have genitals that look typical at birth. Later in life, they may have:
- Irregular menstrual periods, or none at all.
- Trouble getting pregnant.
- Features such as facial hair, more body hair than usual and a deeper voice.
Sometimes, nonclassic CAH may be confused with a hormonal condition that happens during the reproductive years called polycystic ovary syndrome.
Nonclassic CAH symptoms in children of either birth sex also can include:
- Symptoms of early puberty, such as growth of pubic hair sooner than usual.
- Serious acne.
- Rapid growth during childhood with bones that are more developed than is typical.
- Shorter than expected final height.
When to see a doctor
Most often, classic CAH is found at birth through routine newborn screening tests. Or it's found when a baby's outer genitals do not look typical. CAH also may be detected when infants show symptoms of serious illness due to low levels of cortisol, aldosterone or both.
In children who have nonclassic CAH, symptoms of early puberty may appear. If you have concerns about your child's growth or development, schedule a checkup with your child's healthcare professional.
In older people who have irregular periods, trouble getting pregnant or both, screening for CAH may be appropriate.
If you are planning pregnancy or are pregnant and may be at risk of CAH, ask your healthcare professional about genetic counseling. A genetic counselor can tell you if your genes might affect you or any children you decide to have.
Causes
The most common cause of CAH is the lack of the enzyme protein known as 21-hydroxylase. Sometimes, CAH is called 21-hydroxylase deficiency. The body needs this enzyme to make proper amounts of hormones. Very rarely, a lack of other much rarer enzymes also can cause CAH.
CAH is a genetic condition. That means it's passed from parents to children. It's present at birth. Children with the condition have two parents who both carry the genetic change that causes CAH. Or they have two parents who have CAH themselves. This is known as the autosomal recessive inheritance pattern.
People can carry the CAH gene and not have symptoms of the condition. This is called being a silent carrier. If a silent carrier becomes pregnant, that person can pass the gene to a child. If tests show that you're a silent carrier of the CAH gene and you have a partner of the opposite sex, talk with your healthcare professional. It's likely that your partner will need to get tested for the CAH gene before pregnancy so that you can better understand the risks.
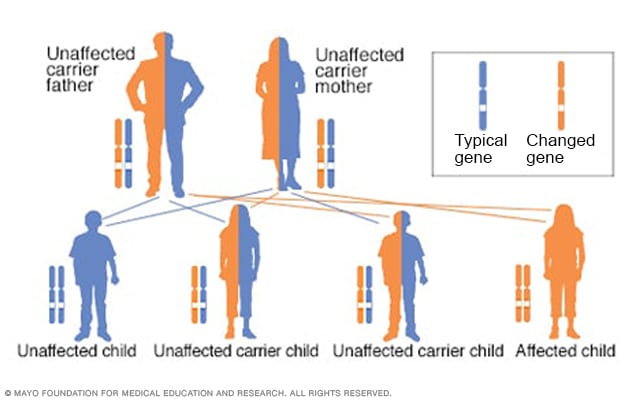
To have an autosomal recessive disorder, you inherit two changed genes, sometimes called mutations. You get one from each parent. Their health is rarely affected because they have only one changed gene. Two carriers have a 25% chance of having an unaffected child with two unaffected genes. They have a 50% chance of having an unaffected child who also is a carrier. They have a 25% chance of having an affected child with two changed genes.
Risk factors
Factors that raise the risk of having CAH include:
- Parents who both have CAH.
- Parents who are both carriers of the changed gene that causes CAH.
- Being of Ashkenazi Jewish, Latino, Mediterranean, Yugoslav or Yup'ik descent.
Complications
People who have classic CAH are at risk of a life-threatening condition called adrenal crisis. This emergency needs to be treated right away. Adrenal crisis can happen within the first few days after birth. It also can be triggered at any age by an infectious illness or physical stress such as surgery.
With adrenal crisis, very low levels of cortisol in the blood can cause:
- Diarrhea.
- Vomiting.
- Dehydration.
- Confusion.
- Low blood sugar levels.
- Seizures.
- Shock.
- Coma.
Aldosterone also may be low. This leads to dehydration, low sodium and high potassium levels. The nonclassic form of CAH doesn't cause adrenal crisis.
People who have either classic or nonclassic CAH may have irregular menstrual cycles and fertility issues.
Prevention
There is no known way to prevent CAH. If you're thinking of starting a family and you're at risk of having a child with CAH, talk with your healthcare professional. You may be told to see a genetic counselor.
Diagnosis
Healthcare professionals may find congenital adrenal hyperplasia (CAH):
- Before a baby is born.
- Shortly after birth.
- During childhood or later in life.
Prenatal testing
Tests used to find CAH before birth in fetuses who are at risk for the condition include:
- Amniocentesis. This procedure involves using a needle to remove a sample of the fluid from the womb. This is called amniotic fluid. Then a lab checks the cells in the fluid.
- Chorionic villus sampling. This test involves removing cells from the organ that provides a fetus with oxygen and nutrients. This organ is called the placenta. A lab checks the sample of placenta cells.
Tests to confirm whether a baby has CAH are done after the baby is born.
Newborns and infants
In the United States and many other countries, newborns are routinely tested for 21-hydroxylase deficiency. The screening test is recommended during the first few days of life. This test can find the classic form of CAH. It doesn't identify the nonclassic form.
In female infants whose outer genitals look very different than is typical, other tests can be done. The tests check structures inside cells that contain genes, called chromosomes, to identify genetic sex. Also, an ultrasound of the pelvis can find the presence of reproductive organs such as the uterus and ovaries.
Children and adults
Tests to find CAH in children and adults include:
- Physical exam. A physical exam usually includes a check of blood pressure and heart rate. Symptoms also are reviewed. If a healthcare professional suspects CAH, blood and urine tests are done.
- Blood and urine tests. These tests look for hormones made by the adrenal glands at levels outside the standard ranges. The tests also check the levels of minerals called electrolytes, such as sodium. These minerals balance the amount of water in the body.
- X-ray. This test might be done to find out if a child's bones are more developed than is typical for the child's age.
- Genetic testing. Genetic testing may be needed to confirm if CAH is the cause of symptoms.
Treatment
For children, a healthcare professional likely will make a referral to a specialist in childhood hormonal issues. This specialist is called a pediatric endocrinologist. For adults, a referral often is made to an adult endocrinologist. The treatment team also may include other healthcare professionals such as:
- A doctor who finds and treats urinary tract conditions, called a urologist.
- A mental health professional called a psychologist.
- A doctor who finds and treats conditions of the female reproductive system, called a reproductive endocrinologist.
- An expert in genes called a geneticist.
Treatment may include medicines, surgery and mental health support.
Medications
The goal of treating CAH with medicines is to lower the amount of androgens the body makes and replace hormones the body lacks. People with classic CAH can manage the condition by taking hormone replacement medicines throughout their lives.
People with nonclassic CAH may not need treatment. Or they may need only small doses of medicines called corticosteroids.
Medicines for CAH are taken every day. During illnesses or times of serious stress, other medicines or higher doses may be needed.
Medicines may include:
- Corticosteroids to replace cortisol.
- Mineralocorticoids to replace aldosterone to help keep salt in the body and get rid of extra potassium.
- Salt supplements to help keep salt in the body.
Regular checkups are needed to make sure the medicines work well. These appointments usually include:
- A physical exam. This exam includes checking a child's growth and development. That involves closely tracking changes in height, weight, blood pressure and bone growth. People with CAH need health checkups on a regular basis throughout their lives.
- Checking for side effects. Medicine side effects may include the loss of bone mass and growth that is slower than usual. The risk of those side effects rises if steroid-type replacement medicine doses are high and used long term.
-
Blood tests to check hormone levels. It's important to have regular blood tests to make sure that hormone levels are balanced. Children who haven't yet reached puberty need enough cortisone to suppress androgens to grow to a typical height. For females with CAH, androgens are suppressed to minimize symptoms such as a deeper voice or extra body hair.
But too much cortisone can cause a condition called Cushing syndrome. Cushing syndrome can lead to symptoms such as a fatty lump between the shoulders and a rounded face. It also can cause high blood pressure, bone loss and type 2 diabetes.
With classic CAH, it's a good idea to wear a medical identification bracelet or necklace that says you have congenital adrenal hyperplasia. It can help a healthcare team provide the right treatment in case of an emergency.
Reconstructive surgery
Some female infants with classic CAH have outer genitals that look very different than is typical. The healthcare team may suggest reconstructive surgery as part of treatment. Surgery can help the genitals function better and look more typical.
Surgery may involve making the clitoris smaller and rebuilding the vaginal opening. The surgery typically is done between about 3 and 6 months of age. Females who have reconstructive genital surgery as infants may need more cosmetic surgery later in life.
Some parents choose to wait to decide on genital surgery for their child. They might delay surgery until the child is old enough to understand the risks and make choices about surgery.
A decision about the timing of surgery should be made after a thorough discussion between the family and the healthcare team.
Mental health support
Mental health support is key for children and adults with CAH. It can help with the social and emotional parts of the condition. Look for a mental health professional who has experience helping people with CAH.
Research
Treatment of CAH during pregnancy with lab-made corticosteroids that cross the placenta to the fetus are controversial and considered experimental. More research is needed to determine the long-term safety and the effect of this treatment on a baby's brain.
Coping and support
Early and steady support from family and healthcare professionals is important. This support can help people with CAH have healthy self-esteem and a satisfying social life. You might want to take these steps:
- Include mental health counseling in a treatment plan as needed.
- Seek help from a mental health professional if you have trouble coping.
Preparing for an appointment
You may start by seeing your family healthcare professional or your child's pediatrician. You may be referred to a specialist trained in finding and treating conditions related to the adrenal glands. This specialist is called an endocrinologist.
Here's some information to help you prepare for your appointment. You may want to take a family member or friend along for support and to help you remember information.
What you can do
To prepare for your appointment:
- Find out if you or your child needs to do anything before the appointment. That might include changing what you or the child eats or drinks to get ready for blood and urine tests.
- Make a list of any symptoms you or your child has had, and for how long.
- Make a list of key medical information. Include recent illnesses, any medical conditions, and the names and dosages of any medicines, vitamins, herbs or other supplements.
- Prepare questions you want to ask your healthcare professional.
Some basic questions to ask may include:
- What is likely causing the symptoms?
- Are there other possible causes for these symptoms?
- What kinds of tests are needed?
- What treatment approach do you recommend?
- What are the expected results of treatment?
- What are the possible side effects of treatment?
- How will you monitor health over time?
- What is the risk of long-term medical problems?
- Do you recommend mental health counseling?
- Do you recommend that our family meet with a genetic counselor?
Feel free to ask any other questions during your appointment.
What to expect from your doctor
Your healthcare professional is likely to ask you questions such as:
- What are your symptoms?
- When did you first start to notice these symptoms?
- Does anyone in your family have congenital adrenal hyperplasia? If so, do you know how it was treated?
Be ready to answer questions so you have time to go over points you want to focus on.
Last Updated Mar 22, 2024
© 2024 Mayo Foundation for Medical Education and Research (MFMER). All rights reserved. Terms of Use




